Raltegravir
(3)


Preço
Tipo de receita
- Branca Comum (Venda Sob Prescrição Médica)
Classe terapêutica
- Outros Antivirais HIV
Forma farmacêutica
- Comprimido mastigável
- Comprimido revestido
Categoria
- HIV/AIDS
- Medicamentos
- Sistema Imunológico
Dosagem
- 25mg
Fabricante
- MSD
Princípio ativo
- Raltegravir
Tipo do medicamento
- Novo
Quantidade
- 60 Unidades




Bula do Raltegravir
Raltegravir, para o que é indicado e para o que serve?
Raltegravir é indicado em combinação com outros agentes antirretrovirais para o tratamento de infecção por HIV-1.
Quais as contraindicações do Raltegravir?
Raltegravir é contraindicado para pacientes com hipersensibilidade conhecida a qualquer componente deste produto.
Tipo de receita
Como usar o Raltegravir?
Raltegravir está disponível como uma formulação de comprimidos de 400 mg e uma formulação de comprimidos mastigáveis nas concentrações de 100 mg (sulcado) e 25 mg.
A dose máxima do comprimido mastigável é de 300 mg 2x/dia. Como as formulações não são bioequivalentes, não substitua os comprimidos mastigáveis pelo comprimido de 400 mg.
Raltegravir pode ser administrado com ou sem alimentos.
Raltegravir deve ser administrado em um regime de combinação com outros agentes antirretrovirais. Para o tratamento de pacientes com infecção por HIV-1, a posologia de Raltegravir é a seguinte:
Adultos:
Um comprimido de 400 mg duas vezes ao dia, por via oral.
Crianças e adolescentes:
12 anos de idade ou mais:
Um comprimido de 400 mg duas vezes ao dia, por via oral.
6 a 11 anos de idade (2 opções posológicas):
- Um comprimido de 400 mg duas vezes ao dia, por via oral (se tiver pelo menos 25 kg de peso) ou
- Comprimidos mastigáveis: peso baseado na dose máxima de 300 mg, duas vezes ao dia, conforme especificado na tabela abaixo.
2 a 5 anos de idade:
Comprimidos mastigáveis: peso baseado na dose máxima de 300 mg, duas vezes ao dia, conforme especificado na tabela abaixo.
Dose Recomendada para Comprimidos Mastigáveis de Raltegravir em Pacientes Pediátricos 2 a 11 Anos de Idade
| Peso Corporal (kg) | Dose | Número de Comprimidos Mastigáveis por dose |
| 7 a < 10 | 50 mg 2x/dia | 0,5 x 100 mg* |
| 10 a < 14 | 75 mg 2x/dia | 3 x 25 mg |
| 14 a < 20 | 100 mg 2x/dia | 1 x 100 mg |
| 20 a < 28 | 150 mg 2x/dia | 1.5 x 100 mg* |
| 28 a < 40 | 200 mg 2x/dia | 2 x 100 mg |
| Pelo menos 40 | 300 mg 2x/dia | 3 x 100 mg |
* O comprimido mastigável de 100 mg pode ser dividido em metades iguais.
Comprimido de 400 mg: Este medicamento não deve ser partido, aberto ou mastigado.
Quais as reações adversas e os efeitos colaterais do Raltegravir?
Adultos
Reações Adversas Em Pacientes Já Tratados
A avaliação da segurança de Raltegravir baseou-se nos relatos de eventos adversos em pacientes que já tratados desde os estudos clínicos randômicos P018 e P019 nos quais foi utilizada a dose recomendada de Raltegravir, 400 mg duas vezes ao dia, em combinação com o Tratamento Otimizado de Base (OBT) em 462 pacientes, em comparação com 237 pacientes que receberam placebo em combinação com OBT. Durante o tratamento duplo-cego, o acompanhamento total foi de 1051 pacientes-anos do grupo recebendo Raltegravir 400 mg 2x/dia e 322 pacientes-anos do grupo recebendo placebo.
Entre os pacientes do grupo de tratamento com Raltegravir 400 mg 2x/dia + OBT (acompanhamento médio de 118,7 semanas) e do grupo para comparação, no qual foi administrado placebo + OBT (acompanhamento médio de 71,0 semanas), na análise agrupada para os estudos P018 e P019, os eventos adversos clínicos mais comumente relatados (>10% em qualquer um dos grupos) de todas as intensidades e independentemente da causalidade foram: diarreia em 26,6% e 24,9%, náuseas em 13,6% e 16,0%, cefaleia em 12,1% e 13,5%,nasofaringite em 14,3% e 8,9%, fadiga em 12,1% e 5,9%, infecção do trato respiratório superior em 15,8% e 10,1%, bronquite em 12,1% e 6,8%, pirexia em 9,7% e 13,9%,vômitos em 8,9% e 11,0% dos pacientes, respectivamente. Nessa análise agrupada, a taxa de descontinuação do tratamento em razão de eventos adversos (clínicos e laboratoriais) foi de 4,5% entre os pacientes que receberam Raltegravir + OBT e de 5,5% entre os pacientes que receberam placebo + OBT.
Eventos Adversos Relacionados ao Medicamento
Os eventos adversos clínicos listados abaixo foram considerados moderados a graves pelos pesquisadores. A causa de tais eventos pode ser atribuída a Raltegravir ou placebo apenas ou em combinação com OBT:
Os eventos adversos de intensidade moderada a grave relacionados ao medicamento que ocorreram em ≥2% dos pacientes adultos que já receberam tratamento em qualquer um dos grupos de tratamento são apresentados na tabela a seguir.
Porcentagem de Pacientes com Eventos Adversos de Intensidade Moderada a Grave e Relacionados ao Medicamento* que Ocorreram em ≥2% dos Pacientes Adultos Já Tratados em Qualquer Grupo de Tratamento**
|
Classe de Órgão Sistêmico |
Estudos Randômicos P018 e P019 | |
| Raltegravir 400 mg 2x/dia + OBT N = 462 |
Placebo |
|
| Acompanhamento Médio (Semanas) 118,7% |
Acompanhamento Médio |
|
|
Distúrbios Gastrintestinais |
||
|
Diarreia |
1,5 |
2,1 |
|
Distúrbios do Sistema Nervoso |
||
| Cefaleia | 2,2% | 0,4% |
* Inclui eventos adversos possível, provável ou muito provavelmente relacionados ao medicamento.
**N = número total de pacientes por grupo de tratamento.
Os eventos adversos clínicos moderados a graves relacionados ao medicamento, que ocorreram em menos de 2% dos pacientes já tratados (n= 462) e que haviam recebido Raltegravir + OBT são listados abaixo, por classe de órgão sistêmico.
[Comuns (≥1/100, <1/10), Incomuns (≥1/1,000, <1/100)].
Distúrbios Cardíacos:
Incomuns:
Extrassístoles ventriculares.
Distúrbios do Ouvido e Labirinto:
Incomuns:
Distúrbios Oculares:
Incomuns:
Comprometimento visual.
Distúrbios Gastrintestinais:
Comuns:
Diarréia, náuseas.
Incomuns:
Dor abdominal, distensão abdominal, dor abdominal alta, vômitos, constipação, desconforto abdominal, dispepsia, flatulência, gastrite, doença de refluxo gastroesofágico, boca seca, eructação.
Distúrbios Gerais e Condições no Local da Administração:
Comuns:
Astenia, fadiga.
Incomuns:
Pirexia, calafrios, edema de face, edema periférico.
Distúrbios Hepatobiliares:
Incomuns:
Distúrbios do Sistema Imunológico:
Incomuns:
Hipersensibilidade ao medicamento.
Infecções e Infestações:
Incomuns:
Herpes simplex, herpes genital, gastrenterite.
Investigação:
Incomuns:
Aumento de peso, redução de peso.
Distúrbios Metabólicos e Nutricionais:
Incomuns:
Diabetes mellitus, dislipidemia, aumento do apetite, redução do apetite.
Distúrbios Musculoesqueléticos e do Tecido Conjuntivo:
Incomuns:
Artralgia, mialgia, lombalgia, dor musculoesquelética, osteoporose, poliartrite.
Distúrbios do Sistema Nervoso:
Incomuns:
Tontura, neuropatia periférica, parestesia, sonolência, cefaleia tensional, tremor.
Distúrbios Psiquiátricos:
Incomuns:
Depressão, insônia, ansiedade.
Distúrbios Renais e Urinários:
Incomuns:
Nefrite, nefrolitíase, noctúria, insuficiência renal, nefrite tubulointersticial.
Distúrbios do Sistema Reprodutivo e da Mama:
Incomuns:
Ginecomastia.
Distúrbios Respiratórios, Torácicos e do Mediastino:
Incomuns:
Epístaxe.
Distúrbios da Pele e do Tecido Subcutâneo:
Incomuns:
Lipodistrofia adquirida, erupção cutânea, hiper-hidrose, dermatite acneiforme, eritema, lipo-hipertrofia, sudorese noturna, erupção cutânea macular, erupção cutânea maculopapular, erupção cutânea prurítica, xerodermia, prurigo, lipoatrofia, prurido.
Eventos Graves
Relacionados ao Medicamento:
Nos estudos clínicos foram relatados os seguintes eventos adversos clínicos graves relacionados ao medicamento: gastrite, hepatite, insuficiência renal, herpes genital, superdosagem acidental.
Eventos adversos em pacientes nunca tratados anteriormente
A avaliação a seguir da segurança de Raltegravir em pacientes nunca expostos a tratamento é baseada no estudo randomizado duplo-cego controlado com agente ativo de pacientes nunca tratados anteriormente, protocolo 021 (STARTMRK) com Raltegravir 400 mg 2x/dia em combinação com uma dose fixa de entricitabina 200 mg (+) tenofovir 245 mg, (N=281) versus efavirenz (EFV) 600 mg à noite antes de se deitar em combinação com entricitabina (+) tenofovir (N=282).
Durante o tratamento duplo-cego, o acompanhamento total para pacientes com Raltegravir 400 mg 2x/dia + entricitabina (+) tenofovir foi de 830 paciente-anos e 788 paciente-anos para pacientes com efavirenz 600 mg à noite antes de se deitar + entricitabina (+) tenofovir.
Os números (%) de pacientes com eventos adversos clínicos e com eventos adversos relacionados ao medicamento no grupo recebendo Raltegravir, foram menos frequentes que no grupo recebendo efavirenz com base nos valores de p nominais (0,109 e <0,001, respectivamente).
Neste estudo, as taxas de descontinuação da terapia devido a eventos adversos (clínicos e laboratoriais) foram de 4,6% em pacientes recebendo Raltegravir + entricitabina (+) tenofovir e 8,5% em pacientes recebendo efavirenz + entricitabina (+) tenofovir.
Para pacientes do grupo recebendo Raltegravir 400 mg 2x/dia + entricitabina (+) tenofovir e do grupo recebendo o agente comparador, efavirenz 600 mg à noite antes de se deitar + entricitabina (+) tenofovir, os eventos adversos clínicos mais comumente relatados (>10% em qualquer grupo), de todas as intensidades e independentemente da relação causal são mostrados na tabela abaixo.
Tabela 11: Porcentagem de Indivíduos com os Eventos Adversos Mais Comumente Relatados (>10%) de Todas as Intensidades* e Independentemente da Relação Causal Ocorrendo em Pacientes Adultos Nunca Expostos a Tratamento em Qualquer Grupo de Tratamento
|
Classe de Sistema Orgânico, |
Estudo Randômico P021 |
|
|
|
Raltegravir 400 mg 2x/dia + Entricitabina (+) Tenofovir (n = 281)† % |
Efavirenz 600 mg |
|
Distúrbios Gastrintestinais |
||
| Diarreia | 25,6 | 27,0 |
| Náusea | 16,7 | 14,5 |
| Vômitos | 8,2 | 10,6 |
|
Distúrbios Gerais e Condições no Local de Administração |
||
| Fadiga | 9,3 | 13,5 |
| Pirexia | 15,7 | 13,8 |
|
Infecções e Infestações |
||
| 11,7 | 13,5 | |
|
Nasofaringite |
26,7 | 22,3 |
|
Infecção do trato respiratório superior |
21,4 | 20,2 |
|
Distúrbios Musculoesqueléticos e do Tecido Conjuntivo |
||
| Artralgia | 8,5 | 11,7 |
| Lombalgia | 12,1 | 9,9 |
|
Distúrbios do Sistema Nervoso |
||
| Tontura | 16,4 | 38,3 |
| Cefaleia | 26,0 | 28,4 |
|
Distúrbios Psiquiátricos |
||
| Sonhos anormais | 8,2 | 13,1 |
| Ansiedade | 8,9 | 13,1 |
| Depressão | 10,3 | 11,7 |
| Insônia | 15,7 | 14,9 |
|
Distúrbios Respiratórios, Torácicos e do Mediastino |
||
| Tosse | 16,7 | 12,1 |
|
Distúrbios da Pele e do Tecido Subcutâneo |
||
|
Erupção cutânea |
7,8 | 13,8 |
* As intensidades são definidas a seguir: leve (consciência do sinal ou sintoma, mas facilmente tolerada); moderada (desconforto suficiente para causar interferência nas atividades usuais); grave (incapacitante com incapacidade de trabalhar ou realizar as atividades usuais).
†n = número total de indivíduos por grupo de tratamento.
Eventos do Sistema Nervoso Central (SNC)
Em pacientes nunca expostos a tratamento (P021) os eventos adversos do sistema nervoso central (SNC), conforme medido pela proporção de pacientes com 1 ou mais sintomas do SNC (descritos abaixo), foram relatados significativamente com menos freqüência no grupo de Raltegravir + entricitabina (+) tenofovir em comparação com o grupo efavirenz + entricitabina (+) tenofovir, p <0,001 , <0,001 e <0,001 para eventos cumulativos até as Semanas 8, 48 e 96, respectivamente.
No grupo Raltegravir, a porcentagem de pacientes com 1 ou mais sintomas do SNC foi de 20,3% em comparação com 52,1% no grupo de efavirenz na Semana 8, e 26,3% em comparação com 58,5% na Semana 48 e 28,8% em comparação com 60,6% na Semana 96. Os eventos adversos do SNC para esta análise foram tontura, insônia, dificuldade de concentração, sonolência, depressão, pesadelos, estado confusional, idéias suicidas, distúrbio do sistema nervoso, distúrbio psicótico, sonhos anormais, tentativa de suicídio, psicose aguda, delírio, nível de consciência deprimido, alucinação, alucinação auditiva, consumação de suicídio e depressão maior.
Eventos Adversos Relacionados ao Medicamento
As reações adversas clínicas listadas abaixo foram consideradas pelos investigadores como sendo de intensidade moderada a grave e a relação causal com Raltegravir/efavirenz apenas ou em combinação com entricitabina (+) tenofovir.
As reações adversas clínicas relacionadas ao medicamento de intensidade moderada a grave ocorrendo em ≥2% dos pacientes adultos nunca tratados anteriormente com qualquer grupo de tratamento são apresentadas na tabela abaixo.
Porcentagem de Pacientes com Eventos Adversos Relacionados ao Medicamento* de Intensidade Moderada a Grave Ocorrendo em ≥2% dos Pacientes Adultos Nunca Tratados Anteriormente em Qualquer Grupo de Tratamento**

Os eventos adversos clínicos relacionados ao medicamento, que ocorreram em menos de 2% dos pacientes nunca tratados anteriormente (n=281) que receberam Raltegravir + entricitabina (+) tenofovir e de intensidade moderada a grave estão listados abaixo por Classe de Sistema Orgânico.
[Comum (≥1/100, <1/10), Incomum (≥1/1.000, <1/100)].
Distúrbios do Sangue e Sistema Linfático:
Incomum:
Dor no linfonodo, neutropenia, anemia, linfadenopatia
Distúrbios do Ouvido e Labirinto:
Incomum:
Tinido, vertigem
Distúrbios Gastrintestinais:
Comum:
Diarreia, dor abdominal.
Incomum:
Vômitos, dor abdominal superior, dispepsia, duodenite erosiva, doença de refluxo gastroesofágico, distensão abdominal.
Distúrbios Gerais e Condições no Local da Administração:
Comum:
Fadiga, astenia.
Incomum:
Massa submandibular.
Distúrbios Hepatobiliares:
Incomum:
Distúrbios do Sistema Imune:
Incomum:
Síndrome de reconstituição imune.
Infecções e Infestações:
Incomum:
Herpes zoster, gastrenterite, foliculite, abscesso de linfonodo.
Distúrbios Metabólicos e Nutricionais:
Incomum:
Redução do apetite, hipercolesterolemia.
Distúrbios Músculo-Esqueléticos e do Tecido Conjuntivo:
Incomum:
Artrite, dor no pescoço
Distúrbios do Sistema Nervoso:
Comum:
Tontura.
Incomum:
Hipersonia, sonolência, comprometimento da memória.
Distúrbios Psiquiátricos:
Comum:
Anormalidade no padrão de sonhos, pesadelos.
Incomum:
Ansiedade, distúrbio mental, estado confusional, depressão, depressão maior.
Distúrbios Renais e Urinários:
Comum:
Nefrolitíase.
Distúrbios do Sistema Reprodutivo e da Mama:
Incomum:
Distúrbios da Pele e Tecido Subcutâneo:
Incomum:
Acne, alopecia, lesão cutânea, lipoatrofia.
Eventos Graves
Os seguintes eventos adversos graves relacionados ao medicamento foram relatados no estudo clínico, P021 em pacientes nunca tratados anteriormente que receberam Raltegravir + entricitabina (+) tenofovir: anemia, síndrome de reconstituição imunológica, distúrbio mental, tentativa de suicídio.
Eventos adversos selecionados
Foram observados casos de câncer em pacientes já tratados anteriormente que iniciaram tratamento com Raltegravir ou placebo, ambos com OBT, e em pacientes nunca expostos ao tratamento que iniciaram tratamento com Raltegravir ou efavirenz, ambos com entricitabina (+) tenofovir; vários eram recidivantes. Os tipos e taxas de câncer específicos foram os esperados em uma população altamente imunodeficiente (muitos apresentavam contagens de CD4+ abaixo de 50 células/mm3 e a maioria apresentava diagnóstico anterior de AIDS). O risco de desenvolvimento de câncer nestes estudos foi similar no grupo que recebeu Raltegravir e no grupo que recebeu o agente comparador.
Foram observadas anormalidades laboratoriais de creatina quinase Grau 2-4 em indivíduos tratados com Raltegravir (veja Tabela 5). Miopatia e rabdomiólise foram relatadas. Utilizar com cautela em pacientes com risco aumentado de miopatia ou rabdomiólise, como pacientes recebendo medicamentos concomitantes conhecidos por causar estas condições.
Erupção cutânea ocorreu mais comumente em pacientes já tratados anteriormente recebendo regimes contendo Raltegravir + darunavir em comparação com pacientes recebendo Raltegravir sem darunavir ou darunavir sem Raltegravir. No entanto, erupção cutânea que foi considerada relacionada ao medicamento ocorreu em taxas similares para todos os três grupos. Estas erupções cutâneas foram de gravidade leve a moderada e não limitaram a terapia; não houve nenhuma descontinuação devido à erupção cutânea. Erupção cutânea ocorreu menos comumente em pacientes nunca tratados anteriormente recebendo Raltegravir em comparação com efavirenz, cada qual em combinação com entricitabina (+) tenofovir.
Pacientes com Condições Coexistentes
Pacientes Coinfectados pelo Vírus da Hepatite B e/ou Hepatite C
Nos estudos Fase III, os pacientes já tratados anteriormente (N=114/699 ou 16%) e os pacientes nunca tratados anteriormente (N = 34/563 ou 6%) com coinfecção crônica (porém não aguda) ativa por hepatite B e/ou hepatite C poderiam ser admitidos desde que os testes de função hepática no baseline não excederam 5 vezes o limite superior do normal. Em geral, o perfil de segurança de Raltegravir em pacientes com coinfecção por hepatite B e/ou hepatite C foi similar ao de pacientes sem coinfecção por hepatite B e/ou hepatite C, embora as taxas de anormalidades de AST e ALT foram um pouco maiores no subgrupo com coinfecção por hepatite B e/ou hepatite C para ambos os grupos de tratamento.
Eventos adversos pediátricos
2 a 18 anos de idade
Raltegravir (substância ativa deste medicamento) foi estudado em 126 crianças e adolescentes de 2 a 18 anos de idade infectados com HIV-1 já tratados com antirretrovirais, em combinação com outros antiretrovirais no IMPAACT P1066. Dos 126 pacientes, 96 receberam a dose recomendada de Raltegravir. Nessas 96 crianças e adolescentes, a frequência, tipo e gravidade das reações adversas relacionadas ao medicamento até a 24a semana foram comparáveis aos observados em adultos.
Um paciente apresentou reações adversas clínicas relacionadas ao medicamento de hiperatividade psicomotora Grau 3, comportamento anormal e insônia; um paciente apresentou uma erupção adversa grave Grau 2 relacionada ao medicamento.
Um paciente apresentou anormalidades laboratoriais relacionadas ao medicamento, AST Grau 4 e ALT Grau 3, as quais foram consideradas graves.
Experiência Pós-comercialização
Os seguintes eventos adversos adicionais foram relatados na experiência pós-comercialização independentemente da relação causal:
Distúrbios do Sangue e Sistema Linfático:
Trombocitopenia.
Distúrbios Hepatobiliares:
Insuficiência hepática (com e sem hipersensibilidade associada) em pacientes com doença hepática subjacente e/ou medicações concomitantes.
Distúrbios Musculoesqueléticos e do Tecido Conjuntivo:
Rabdomiólise.
Distúrbios do Sistema Nervoso:
Ataxia cerebelar.
Distúrbios Psiquiátricos:
Depressão (particularmente em pacientes com histórico preexistente de doença psiquiátrica), incluindo idéias e comportamentos suicidas.
Distúrbios da Pele e Tecido Subcutâneo:
Síndrome de Stevens-Johnson, erupção cutânea com eosinofilia e sintomas sistêmicos (DRESS)
Achados De Exames Laboratoriais
Anormalidades Laboratoriais:
Nos estudos P018 e P019, as porcentagens de pacientes adultos já tratados anteriormente que receberam Raltegravir 400 mg duas vezes ao dia ou placebo (ambos com OBT) com anormalidades laboratoriais selecionadas, de grau 2 a 4, que representam agravamento de Grau em relação ao período basal são apresentadas na tabela a seguir.
Anormalidades Laboratoriais Selecionadas, de Grau 2 a 4, em Pacientes Já Tratados
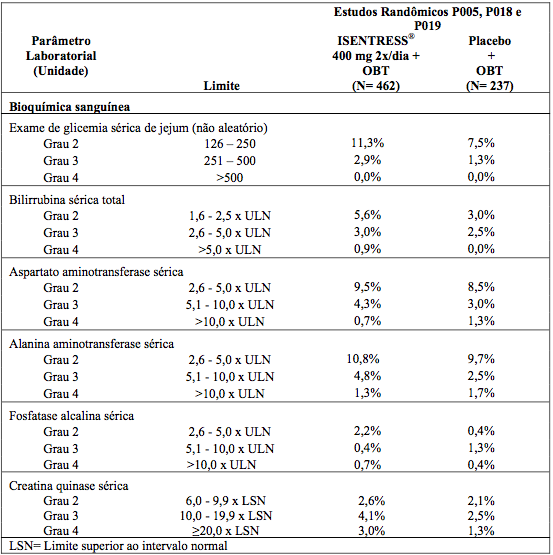
As porcentagens de pacientes adultos sem tratamento anterior recebendo tanto Raltegravir 400mg duas vezes ao dia ou efavirenz (ambos com entricitabina (+) tenofovir), no estudo P021 com anormalidades laboratoriais selecionadas de Grau 2 a 4 que representam um pior Grau do parâmetro inicial estão apresentados na Tabela abaixo.
Anormalidades Laboratoriais Selecionadas de Grau 2 a 4 Relatadas em Pacientes Não Tratados Anteriormente

Lipídios, Alteração em relação ao período basal
Para P021, alterações em relação ao período basal em testes de lipídios em jejum são mostradas na Tabela a seguir.
Valores de Lipídios em P021, Alteração em Relação ao Período Basal em Lipídios Séricos na Semana 144

Atenção: Este produto é um medicamento que possui nova forma farmacêutica e nova concentração no país e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Interação medicamentosa: quais os efeitos de tomar Raltegravir com outros remédios?
O raltegravir não é um substrato das enzimas do citocromo P450 (CYP) e não inibe (CI50>100 μM) a CYP1A2, a CYP2B6, a CYP2C8, a CYP2C9, a CYP2C19, a CYP2D6 ou a CYP3A in vitro. Além disso, in vitro, o raltegravir não induziu a CYP3A4. Um estudo de interação medicamentosa com midazolam confirmou a baixa propensão do raltegravir para alterar a farmacocinética dos agentes metabolizados pela CYP3A4 in vivo pela demonstração da ausência de efeito significativo do raltegravir sobre a farmacocinética do midazolam, um substrato sensível à CYP3A4.
Da mesma maneira, o raltegravir não é um inibidor (CI50>50 μM) das UDP-glicuronosiltransferases (UGTs) avaliadas (UGT1A1, UGT2B7) e não inibe o transporte mediado pela P-glicoproteína. Com base nesses dados, não se espera que Raltegravir afete a farmacocinética dos medicamentos substratos dessas enzimas ou da P-glicoproteína (por exemplo, inibidores da protease, ITRNNs, metadona, analgésicos opioides, vastatinas, antifúngicos azois, inibidores da bomba de prótons, e agente[s] para o tratamento da disfunção erétil).
Com base nos estudos in vivo e in vitro, o raltegravir é eliminado principalmente pelo metabolismo via glicuronidação mediada pela UGT1A1.
A co-administração de Raltegravir com medicamentos que são potentes indutores da UGT1A1, como a rifampicina (indutor de várias enzimas metabolizantes de fármacos), reduz as concentrações plasmáticas de Raltegravir. Deve-se ter cuidado ao se coadministrar Raltegravir com rifampicina ou outros fortes indutores da UGT1A1. O impacto de outros potentes indutores de enzimas metabolizadoras de fármacos, como fenitoína e fenobarbital, sobre a UGT1A1 é desconhecido. Outros indutores menos potentes (por exemplo, efavirenz, nevirapina, rifabutina, glicocorticoides, erva-de-são- joão, pioglitazona) podem ser utilizados com a dose recomendada de Raltegravir.
A coadministração de Raltegravir com medicamentos conhecidos por serem potentes indutores da UGT1A1 (por exemplo, atazanavir) aumenta os níveis plasmáticos de Raltegravir. No entanto, o aumento é discreto e o tratamento combinado com esses inibidores foi bem tolerado nos estudos clínicos, de forma que nenhum ajuste de dose é necessário.
A coadministração de Raltegravir com medicamentos conhecidos por aumentarem o pH gástrico (p.ex., omeprazol) pode aumentar os níveis plasmáticos de Raltegravir com base no aumento da solubilidade de Raltegravir em pH mais alto.
Em indivíduos que receberam Raltegravir em combinação com inibidores da bomba de prótons ou bloqueadores de H2 nos Protocolos 018 e 019, foram observados perfis de segurança comparáveis neste subgrupo em relação a indivíduos não recebendo inibidores da bomba de prótons ou bloqueadores da H2. Com base nestes dados, os inibidores da bomba de prótons ou bloqueadores da H2 podem ser coadministrados com Raltegravir sem ajuste de dose.
Efeito do Raltegravir sobre a Farmacocinética de Outros Agentes:
Nos estudos de interação medicamentosa, o raltegravir não apresentou efeitos clinicamente significativos sobre a farmacocinética de contraceptivos hormonais, metadona, tenofovir, midazolam, lamivudina, etravirina, e darunavir/ritonavir. Em um estudo de interação medicamentosa de doses múltiplas, os valores de AUC de etinil estradiol e de norelgestromina foram de 98% e 114%, respectivamente, quando coadministrados com raltegravir em comparação com quando administrados sem raltegravir.
Em um estudo de interação medicamentosa de doses múltiplas, a AUC e as concentrações de vale do tenofovir coadministrado com o raltegravir foram de 90% e 87% dos valores obtidos com a monoterapia com o tenofovir. Em outro estudo de interação medicamentosa, a AUC do midazolam coadministrado foi 92% do valor obtido com o midazolam isoladamente. Em um estudo fase II, a farmacocinética da lamivudina foi semelhante em pacientes que receberam combinações com raltegravir versus com efavirenz.
Efeito de Outros Agentes sobre a Farmacocinética do Raltegravir:
Nos estudos de interação medicamentosa, atazanavir, efavirenz, ritonavir, tenofovir e tipranavir/ritonavir não apresentaram efeito clinicamente significativo sobre a farmacocinética do raltegravir. A rifampicina, forte indutora das enzimas metabolizadoras de medicamentos, causou redução dos níveis de vale do raltegravir. Todos os estudos de interação foram realizados em adultos. As interações medicamentosas encontram-se descritas adicionalmente na tabela abaixo.
Efeito de Outros Agentes sobre a Farmacocinética do Raltegravir

Quais cuidados devo ter ao usar o Raltegravir?
Gravidez:
Categoria de risco - C.
Os estudos de toxicidade ao desenvolvimento foram realizados em coelhos (em doses de até 1.000 mg/kg/dia) e ratos (em doses de até 600 mg/kg/dia). As doses mais altas nestes estudos produziram exposições sistêmicas nessas espécies aproximadamente 3 a 4 vezes acima da exposição à dose recomendada para humanos.
Não foram observadas alterações externas, viscerais ou esqueléticas relacionadas ao tratamento em coelhos. Foram observados aumentos relacionados ao tratamento em relação aos controles na incidência de costelas supranumerárias em ratos na dose de 600 mg/kg/dia (exposições 4,4 vezes acima da exposição à dose humana recomendada). Tanto em coelhos como em ratos, não foram observados efeitos relacionados ao tratamento na sobrevida embrio/fetal ou no peso dos fetos.
Em ratos, a dose administrada a fêmeas prenhes foi de 600 mg/kg/dia, e as concentrações plasmáticas médias do fármaco no plasma de fetos foram aproximadamente 1,5 a 2,5 vezes maiores que no plasma materno 1 hora e 24 horas pós-dose, respectivamente. Em coelhos, uma dose de 1.000 mg/kg/dia foi administrada a fêmeas prenhes, e as concentrações médias do fármaco no plasma fetal foram aproximadamente 2% da concentração materna média tanto 1 como 24 horas após a dose. Estudos toxicocinéticos demonstraram transferência placentária do fármaco em ambas as espécies.
Não existem estudos adequados e bem-controlados em mulheres grávidas; portanto, a segurança de Raltegravir em mulheres grávidas não é conhecida. A exemplo de outros antirretrovirais, não se recomenda o uso de Raltegravir durante a gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação:
Não se sabe se o raltegravir é secretado no leite humano; no entanto, o raltegravir é secretado no leite de ratas lactantes. Em ratas que receberam doses de 600 mg/kg/dia, as concentrações médias do fármaco no leite foram aproximadamente 3 vezes maiores do que no plasma materno.
A amamentação não é recomendada durante o tratamento com Raltegravir. Além disso, recomenda-se que mães infectadas pelo HIV não amamentem seus bebês para evitar o risco de transmissão pós-natal do HIV.
Síndrome de Reconstituição Imunológica:
Durante a fase inicial do tratamento, os pacientes que respondem ao tratamento antirretroviral podem desenvolver resposta inflamatória a infecções oportunistas indolentes ou residuais (como Mycobacterium avium complexo, citomegalovírus, pneumonia por Pneumocystis jiroveci e tuberculose ou reativação do vírus varicella zoster), que podem precisar de avaliação e tratamento adicionais.
Reações Cutâneas e de Hipersensibilidade Graves:
Foram relatadas reações cutâneas graves, potencialmente fatais, e fatais em pacientes usando Raltegravir concomitantemente com outros medicamentos associados a essas reações. Essas reações incluem casos de síndrome Stevens-Johnson e necrólise epidérmica tóxica. Reações de hipersensibilidade também foram relatadas e foram caracterizadas por erupção cutânea, achados constitucionais, e algumas vezes, disfunção orgânica, incluindo insuficiência hepática.
Descontinue Raltegravir e outros agentes suspeitos imediatamente se sinais ou sintomas de reações cutâneas ou de hipersensibilidade graves se desenvolverem (incluindo, porém não se limitando a, erupção cutânea grave ou erupção cutânea acompanhada de febre, mal-estar geral, fadiga, dores musculares ou articulares, bolhas, lesões orais, conjuntivite, edema facial, hepatite, eosinofilia, angioedema).
O status clínico, incluindo as aminotransferases hepáticas, deve ser monitorado e deve-se iniciar o tratamento apropriado. Postergar a interrupção do tratamento com Raltegravir ou outros agentes suspeitos após o início da erupção cutânea grave pode resultar em uma reação potencialmente fatal.
Uso Pediátrico:
A segurança, a tolerabilidade, o perfil farmacocinético e a eficácia de Raltegravir foram avaliados em crianças e adolescentes infectados pelo HIV-1 com 2 a 18 anos de idade em um estudo clínico aberto e multicêntrico, o IMPAACT P1066.
O perfil de segurança foi comparável ao observado em adultos. A segurança e a eficácia de Raltegravir em crianças com menos de 2 anos de idade não foram estabelecidas.
Uso em Idosos:
Estudos clínicos com Raltegravir não incluíram número suficiente de pacientes com 65 anos ou mais para determinar se eles respondem de forma diferente de pacientes mais jovens.
Outras experiências clínicas relatadas não identificaram diferença de resposta entre pacientes idosos e jovens. Em geral, a seleção da dose para um paciente idoso deve ser feita com cautela, refletindo a frequência mais alta de insuficiência hepática, renal ou cardíaca e de doenças concomitantes ou outros tratamentos medicamentosos.
Atenção: o uso incorreto causa resistência do vírus da AIDS e falha no tratamento.
Interações Medicamentosas:
Deve-se ter cuidado ao se co-administrar Raltegravir com fortes indutores da uridina difosfato glicuronosiltransferase (UGT) 1A1 (p. ex., rifampicina) em razão da redução de concentração plasmática do Raltegravir.
Outras advertências:
Atenção fenilcetonúricos: contém fenilalanina
Raltegravir comprimidos mastigáveis contém fenilalanina, um componente do aspartame. Cada comprimido de 25 mg de Raltegravir comprimidos mastigáveis contém cerca de 0,05 mg de fenilalanina. Cada comprimidos de 100 mg de Raltegravir contém cerca de 0,10 mg de fenilalanina. Fenilalanina pode ser perigosa para pacientes que tem fenilcetonúria.
Qual a ação da substância do Raltegravir?
Resultados de eficácia
Adultos
Descrição dos Estudos Clínicos:
As evidências de eficácia durável de Raltegravir baseiam-se nas análises dos dados de 96 semanas de 2 estudos randômicos, em andamento, duplo-cegos e controlados por placebo: BENCHMRK 1 e BENCHMRK 2 (Protocolos 018 e 019), compostos por pacientes adultos infectados por HIV- 1 já tratados com agentes antirretrovirais e na análise dos dados de 156 semanas de um estudo em andamento, randômico, duplo-cego, controlado com fármaco ativo, STARTMRK (P021).
Pacientes Já Tratados
BENCHMRK 1 e BENCHMRK 2 são estudos fase III para avaliar a segurança e a atividade antirretroviral de Raltegravir 400 mg 2x/dia em combinação com um Tratamento Otimizado de Base (OBT), versus a OBT isoladamente, em pacientes infectados pelo HIV, com 16 anos de idade ou mais, com resistência documentada a pelo menos 1 fármaco de cada uma das 3 classes (ITRNs, ITRNNs, IPs) de tratamento antirretroviral. A distribuição randômica foi estratificada por grau de resistência ao IP (1 IP vs. >1 IP) e uso de enfuvirtida na OBT. Antes da distribuição randômica, a OBT foi selecionada pelo pesquisador com base no teste de resistência genotípica/fenotípica e no histórico anterior de ART.
A tabela 1 mostra as características demográficas entre os pacientes do braço Raltegravir 400 mg 2x/dia e os pacientes do braço que recebeu placebo.
Tabela 1: Características no Período Basal
|
BENCHMRK 1 e 2 agrupados |
Raltegravir 400 mg 2xqdia + OBT (N=462) |
Placebo + OBT (N=237) |
|
Sexo n (%) |
||
| Masculino | 405 (87,7) | 210 (88,6) |
| Feminino | 57 (12,3) | 27 (11,4) |
|
Raça n (%) |
||
| Brancos | 301 (65,2) |
173 (73,0) |
| Negros | 65 (14,1) |
26 (11,0) |
| Asiáticos | 16 (3,5) |
6 (2,5) |
| Hispânicos | 53 (11,5) |
19 (8,0) |
| Outros | 27 (5,8) |
13 (5,5) |
|
Idade (anos) |
||
|
Mediana (mín., máx.) |
45,0 (16 a 74) |
45,0 (17 a 70) |
|
Contagem de Células CD4 |
||
|
Mediana (mín, máx), células/mm3 |
119 (1 a 792) |
123 (0 a 759) |
|
≤ 50 células/mm3, n (%) |
146 (31,6) |
78 (32,9) |
|
50 < e ≤ 200 células/mm3, n (%) |
173 (37,4) |
85 (35,9) |
|
RNA de HIV Plasmático |
||
|
Mediana (mín., máx.), log10 cópias/mL |
4,8 (2,3 a 5,9) |
4,7 (2,3 a 5,9) |
|
> 100.000 cópias/mL, n (%) |
165 (35,7) |
78 (32,9) |
|
Histórico de AIDS n (%) |
||
| Sim | 427 (92,4) |
215 (90,7) |
|
Uso Anterior de ART, Mediana (1o trimestre, 3° trimestre) |
||
|
Anos de uso de ART |
10,1 (7,3 a 12,1) |
10,2 (7,9 a 12,4) |
|
Número de ARTs |
12,0 (9 a 15) |
12,0 (9 a 14) |
|
Coinfecção por Hepatite† n (%) |
||
|
Sem Hepatite B ou C |
385 (83,3) | 200 (84,4) |
|
Hepatite B apenas |
36 (7,8) | 7 (3,0) |
|
Hepatite C apenas |
37 (8,0) | 28 (11,8) |
|
Coinfecção por Hepatite B e C |
4 (0,9) | 2 (0,8) |
|
Estrato n (%) |
||
|
Enfuvirtida na OBT |
175 (37,9) |
89 (37,6) |
|
Resistente a ≥ 2 IPs |
447 (96,8) |
226 (95,4) |
†Positivo para antígeno de superfície da hepatite B ou positivo para anticorpo da hepatite C.
A tabela 2 compara as características dos pacientes sob Tratamento Otimizado de Base no período basal que receberam Raltegravir 400 mg 2x/dia versus as características dos pacientes do braço de controle
|
BENCHMRK 1 e 2 agrupados |
Raltegravir 400 mg 2xqdia + OBT (N=462) |
Placebo + OBT |
|
Número de ARTs na OBT |
||
|
Mediana (mín., máx.) |
4,0 (1 a 7) |
4,0 (2 a 7) |
|
Número de IPs Ativos na OBT pelo Teste de Resistência Fenotípica† |
||
| 0 | 165 (35,7) |
96 (40,5) |
| 1 ou mais | 278 (60,2) |
137 (57,8) |
|
Escore de Sensibilidade Fenotípica (ESF)‡ |
||
| 0 | 67 (14,5) |
43 (18,1) |
| 1 | 144 (31,2) |
71 (30,0) |
| 2 | 142 (30,7) |
66 (27,8) |
| 3 ou mais | 85 (18,4) |
48 (20,3) |
|
Escore de Sensibilidade Genotípica (ESG)‡ |
||
| 0 | 116 (25,1) |
65 (27,4) |
| 1 | 177 (38,3) |
95 (40,1) |
| 2 | 111 (24,0) |
49 (20,7) |
| 3 ou mais | 51 (11,0) |
23 (9,7) |
† O uso de darunavir na OBT em pacientes que não haviam recebido darunavir foi contabilizado como um IP ativo.
‡ O Escore de Sensibilidade Fenotípica (ESF) e o Escore de Sensibilidade Genotípica (ESG) foram definidos como o total de ARTs orais na OBT aos quais um isolado viral do paciente apresentou sensibilidade fenotípica e sensibilidade genotípica, respectivamente, com base nos testes de resistência fenotípica e genotípica. O uso de enfuvirtida na OBT de pacientes que não haviam recebido enfuvirtida foi contabilizado como um fármaco ativo na OBT no ESG e no ESF. Da mesma maneira, o uso de darunavir na OBT em pacientes que não haviam recebido darunavir foi contabilizado como um fármaco ativo na OBT.
Os resultados da 48a e da 96a semanas para os 699 pacientes distribuídos de forma randômica e que receberam a dose recomendada de Raltegravir 400 mg 2x/dia dos estudos BENCHMRK 1 e 2 agrupados são mostrados na tabela 3.

O percentual (IC 95%) de pacientes que atingiram RNA de HIV <50 cópias/mL em função do tempo é apresentado na Figura 1.

As respostas virológicas na 96a semana por escore de sensibilidade genotípica e fenotípica no período basal são mostradas na tabela 4.

Troca de Pacientes Suprimidos de Lopinavir/Ritonavir para Raltegravir
Os estudos SWITCHMRK 1 & 2 avaliaram pacientes infectados por HIV recebendo tratamento supressor (RNA do HIV na triagem<50 cópias/mL; regime estável >3 meses) com lopinavir 200 mg/ritonavir 50 mg, 2 comprimidos duas vezes ao dia mais pelo menos 2 inibidores nucleosídeos de transcriptase reversa e os distribuíram de forma randômica na proporção de 1:1 para continuar com lopinavir/ritonavir 2 comprimidos duas vezes ao dia (n=174 e n=178, respectivamente) ou substituir lopinavir/ritonavir por raltegravir 400 mg duas vezes ao dia (n=174 e n=176, respectivamente). Os pacientes com histórico anterior de falha virológica não foram excluídos o número de tratamentos antirretrovirais anteriores não era limitado.
Esses estudos foram encerrados após a análise primária de eficácia na 24a semana porque não conseguiram demonstrar não-inferioridade do raltegravir versus lopinavir/ritonavir. Em ambos os estudos na 24a semana, a supressão do RNA de HIV para menos de 50 cópias/mL foi mantida em 84,4% do grupo raltegravir versus 90,6% do grupo lopinavir/ritonavir, (não completar= falha). Em pacientes que nunca apresentaram falha virológica antes da entrada no estudo, taxas de resposta virológica similares foram observadas nos grupos de raltegravir e de lopinavir/ritonavir.
Pacientes Nunca Expostos a Tratamento
O STARTMRK é um estudo Fase III para avaliar a segurança e a atividade antirretroviral de Raltegravir 400 mg 2x/dia + entricitabina/tenofovir versus efavirenz + entricitabina/tenofovir em pacientes infectados por HIV nunca expostos a tratamento com RNA de HIV >5000 cópias/mL. A distribuição randômica foi estratificada por nível de triagem de RNA de HIV (≤50.000 cópias/mL; e >50.000 cópias/mL) e por status de hepatite.
A tabela 5 mostra as características demográficas entre pacientes do grupo que recebeu Raltegravir 400 mg 2x/dia e pacientes do grupo que recebeu efavirenz.

Os resultados da 48a e 156a semanas do STARTMRK são mostrados na Tabela 6.
Tabela 6: Resultados por Grupo de Tratamento até a 48a e 156a Semanas

A Figura 2 apresenta a proporção de pacientes com RNA de HIV plasmático <50 cópias/mL em função do tempo por grupo de tratamento. Os pacientes tratados com Raltegravir atingiram supressão viral (RNA de HIV <50 cópias/mL) mais cedo que os que receberam EFV. Durante 156 semanas de tratamento, 75% do grupo que recebeu Raltegravir 400 mg 2x/dia e 68% no grupo comparador atingiram RNA de HIV <50 cópias/mL (abordagem NC=F).

Os pacientes que receberam Raltegravir atingiram supressão viral (RNA de HIV <50 cópias/mL) mais cedo que os que receberam efavirenz, ambos em combinação com entricitabina/tenofovir.
No estudo STARTMRK da terapia de combinação antirretroviral em pacientes nunca expostos a tratamento, Raltegravir com entricitabina/tenofovir demonstrou eficácia virológica e imunológica consistente em relação ao efavirenz com entricitabina/tenofovir por meio de dados demográficos e fatores prognósticos basais, incluindo: nível basal de RNA de HIV plasmático >100.000 cópias/mL, células CD4 basais ≤50 células/mm3, grupos demográficos (incluindo idade, sexo, região e raça), com status de coinfecção por hepatites (hepatite B e/ou C) e subtipos virais (comparando clade não B com um grupo clade B).
Observou-se eficácia consistente de Raltegravir em todos os subtipos de HIV, com 88,0% (162/184) e 94,0% (47/50) dos pacientes com subtipos B e não B, respectivamente, atingindo RNA de HIV <50 cópias/mL na 156a semana (abordagem FO).
Durante 144 semanas de terapia, Raltegravir demonstrou efeitos mínimos sobre os níveis lipídicos séricos com pequenos aumentos de colesterol total e de colesterol LDL e redução dos níveis séricos de triglicérides. O grupo tratado com efavirenz apresentou alteração média significativamente maior em relação ao período basal dos níveis de colesterol total, triglicérides, colesterol não HDL, e colesterol LDL. Aumentos modestos de HDL foram observados em ambos os grupos, significativamente maiores para efavirenz.
Pacientes Pediátricos
2 a 18 anos de idade
O estudo IMPAACT P1066 é um estudo aberto e multicêntrico Fase I/II para avaliar o perfil farmacocinético, a segurança, a tolerabilidade e a eficácia de raltegravir em crianças infectadas pelo HIV. Este estudo admitiu 126 crianças e adolescentes de 2 a 18 anos de idade já tratados. Os pacientes foram estratificados por idade, admitindo adolescentes primeiramente e, então, crianças mais jovens, sucessivamente.
Os pacientes receberam a formulação de comprimidos de 400 mg (6 a 18 anos de idade) ou a formulação de comprimidos mastigáveis (2 a 11 anos de idade). O raltegravir foi administrado a um regime de base otimizado.
O estágio de definição da dose inicial incluiu avaliação farmacocinética intensiva. A seleção da dose foi baseada na obtenção de exposição plasmática similar de raltegravir e concentração de vale conforme observado em adultos, e segurança aceitável de curto prazo. Após a seleção da dose, outros pacientes foram admitidos para avaliação da segurança, tolerabilidade e eficácia prolongada. Dos 126 pacientes, 96 receberam a dose recomendada de Raltegravir.
Esses 96 pacientes tinham idade mediana de 13 (faixa de 2 a 18) anos, 51% eram do sexo feminino, 34% caucasianos e 59% negros. No período basal, o nível médio plasmático de RNA de HIV-1 era de 4,3 log10 cópias/mL, a contagem mediana de células CD4, de 481 células/mm3 (intervalo: 0 – 2361) e a % mediana de CD4 era de 23,3% (intervalo: 0 – 44).
No geral, 8% apresentaram níveis plasmáticos basais de RNA de HIV-1 >100.000 cópias/mL e 59% apresentaram classificação clínica de CDC HIV categoria B ou C. A maioria dos pacientes havia utilizado anteriormente pelo menos um ITRNN (78%) ou um IP (83%).
Noventa e três (97%) pacientes com 2 a 18 anos de idade completaram 24 semanas de tratamento (3 descontinuaram por falta de adesão ao tratamento). Na 24a semana, 72% atingiram redução ≥1 log10 RNA de HIV em relação ao período basal ou <400 cópias/mL; 54% atingiram RNA de HIV <50 cópias/mL. O aumento médio da contagem de CD4 (porcentagem) do período basal até a 24a semana foi de 119 células/mm3 (3,8%).
Setenta e dois (95%) pacientes com 6 a 18 anos de idade completaram 48 semanas de tratamento (4 descontinuaram por falta de adesão ao tratamento). Na 48a semana, 77% atingiram redução ≥1 log10 RNA de HIV em relação ao período basal ou <400 cópias/mL; 56% atingiram RNA de HIV <50 cópias/mL. O aumento médio da contagem de CD4 (porcentagem) do período basal até a 48a semana foi de 155 células/mm3 (4,7%).
Características farmacológicas
Raltegravir é um inibidor da integrase, enzima responsável pela transferência do filamento de DNA viral do HIV, ativo contra o vírus da imunodeficiência humana (HIV-1).
Mecanismo de Ação:
O raltegravir inibe a atividade catalítica da integrase do HIV, uma enzima decodificada pelo HIV e necessária para a replicação viral. A inibição da integrase evita a inserção ou integração covalente do genoma do HIV no genoma da célula hospedeira durante a fase inicial da infecção. Os genomas do HIV que não conseguem se integrar, não conseguem dirigir a produção de novas partículas infecciosas virais e dessa forma, a inibição da integração impede a propagação da infecção viral. O raltegravir não inibiu de forma significativa as fosforiltransferases humanas, incluindo as polimerases α, β, e γ do DNA.
Absorção - Adultos:
O raltegravir é rapidamente absorvido em jejum com Tmáx de aproximadamente 3 horas após a dose. A AUC e a Cmáx do raltegravir aumentam de forma proporcional com a dose no intervalo de dose de 100 mg a 1.600 mg. A C12h do raltegravir aumenta proporcionalmente com a dose no intervalo de dose de 100 a 800 mg e aumenta um pouco menos que proporcionalmente com a dose no intervalo de dose de 100 mg a 1.600 mg. Com a administração duas vezes ao dia, o estado de equilíbrio farmacocinético é atingido rapidamente, aproximadamente nos primeiros 2 dias de administração. Existe pouco ou nenhum acúmulo de AUC e de Cmáx e evidências de discreto acúmulo de C12h. A biodisponibilidade absoluta do raltegravir ainda não foi estabelecida.
Em pacientes que recebem monoterapia com 400 mg duas vezes ao dia, a exposição ao raltegravir foi caracterizada por uma AUC0-12h média geométrica de 14,3 μM●h e C12h de 142 nM.
Efeito da Presença de Alimentos sobre a Absorção Oral:
Raltegravir pode ser administrado com ou sem alimentos. O raltegravir foi administrado independentemente da presença de alimentos nos estudos pivotais de segurança e eficácia em pacientes infectados pelo HIV. O efeito do consumo de refeições com baixo, moderado e alto teor de gorduras sobre a farmacocinética de estado de equilíbrio do raltegravir foi determinado em voluntários saudáveis.
A administração de doses múltiplas de raltegravir após uma refeição de teor moderado de gorduras não afetou a AUC do raltegravir em um grau clinicamente significativo com um aumento de 13% em relação ao estado de jejum. A C12hr do raltegravir foi 66% maior e a Cmáx foi 5% maior após uma refeição com teor moderado de gorduras em comparação com o estado de jejum.
A administração de raltegravir após uma refeição com alto teor de gorduras aumentou a AUC e a Cmáx em aproximadamente 2 vezes e aumentou a C12hr em 4,1 vezes. A administração do raltegravir após uma refeição com baixo teor de gorduras diminuiu a AUC e a Cmáx em 46% e a 52%, respectivamente; a C12hr ficou basicamente inalterada. A presença de alimentos parece aumentar a variabilidade farmacocinética em relação ao estado de jejum.
Distribuição - Adultos:
O raltegravir apresenta taxa de ligação a proteínas plasmáticas de aproximadamente 83% no intervalo de concentração de 2 a 10 μM. O raltegravir atravessa prontamente a placenta de ratas, porém não penetrou o cérebro em grau perceptível.
Metabolismo e Eliminação - Adultos:
A meia-vida terminal aparente do raltegravir é de aproximadamente 9 horas, com uma meia-vida de fase α mais curta (~1 hora) respondendo pela paior parte da AUC. após a administração de uma dose oral de raltegravir radiomarcado, aproximadamente 51% e 32% da dose foram excretados nas fezes e urina, respectivamente.
Nas fezes, apenas o raltegravir estava presente, e a maioria provavelmente era derivada da hidrólise do glicuronídeo de raltegravir na bile, conforme observado nas espécies pré-clínicas. Dois componentes, raltegravir e glicuronídeo de raltegravir, foram detectados na urina e responderam por cerca de 9% e 23% da dose, respectivamente.
A principal entidade circulante era o raltegravir, representando aproximadamente 70% da radioatividade total; a radioatividade restante no plasma foi atribuída ao glicuronídeo de raltegravir. Estudos que utilizaram inibidores químicos seletivos da isoforma e as UDP- glicuronosiltransferases (UGT) expressas pelo cDNA mostram que a UGT1A1 é a principal enzima responsável pela formação do glicuronídeo de raltegravir. Assim, os dados indicam que o principal mecanismo de clearance do raltegravir em humanos é a glicuronidação mediada por UGT1A1.
Características em Pacientes
Sexo:
Um estudo sobre a farmacocinética do raltegravir foi realizado em homens e mulheres jovens adultos saudáveis. Além disso, em uma análise composta dos dados farmacocinéticos de 103 indivíduos saudáveis e 28 pacientes com HIV que receberam monoterapia com raltegravir administrada em jejum foi avaliada se haveria alguma diferença de resposta recorrente do sexo. Essa hipótese também foi avaliada em uma análise da farmacocinética populacional (PK) dos dados de concentração de 80 indivíduos saudáveis e pacientes com HIV que recebem apenas raltegravir ou raltegravir em combinação com outros fármacos, na presença ou não de alimentos. Não houve nenhuma diferença farmacocinética clinicamente importante decorrente do sexo. Não é necessário nenhum ajuste de dose.
Idade:
O efeito da idade (18 anos ou mais) sobre a farmacocinética do raltegravir foi avaliado na análise composta e na análise de farmacocinética populacional. Não houve efeito clinicamente significativo decorrente da idade sobre a farmacocinética do raltegravir. Não é necessário ajuste de dose.
Pacientes Pediátricos:
Com base em um estudo de comparação de formulação em voluntários adultos saudáveis, o comprimido mastigável possui maior biodisponibilidade oral em comparação com o comprimido de 400 mg. Neste estudo, a administração do comprimido mastigável com uma refeição com alto teor de gorduras levou à redução média de 6% da AUC, redução de 62% da Cmáx, e aumento de 188% da C12h em comparação com a administração em jejum.
A administração do comprimido mastigável com uma refeição de alto teor de gorduras não afeta a farmacocinética do raltegravir em um grau clinicamente significativo e o comprimido mastigável pode ser administrado independentemente da presença de alimentos.
As doses recomendadas a crianças e adolescentes infectados com HIV com 2 a 18 anos de idade resultou em perfil farmacocinético de raltegravir similar ao observado em adultos recebendo 400 mg duas vezes ao dia. A tabela 7 mostra os parâmetros farmacocinéticos do comprimido de 400 mg (6 a 18 anos de idade) e do comprimido mastigável (2 a 11 anos de idade).

A farmacocinética do raltegravir em crianças com menos de 2 anos não foi estabelecida.
Raça:
O efeito da raça sobre a farmacocinética do raltegravir foi avaliado na análise composta. Não houve efeito clinicamente significativo da raça sobre a farmacocinética do raltegravir. Não é necessário nenhum ajuste de dose.
Índice de Massa Corporal (IMC):
A análise composta avaliou o efeito do IMC na farmacocinética do raltegravir. Não houve efeito clinicamente significativo do IMC na farmacocinética do raltegravir em adultos.
Além disso, nenhum efeito clinicamente significativo do peso corporal na farmacocinética do raltegravir foi identificado na análise da farmacocinética populacional. Não é necessário nenhum ajuste de dose.
Insuficiência Hepática:
O raltegravir é eliminado principalmente por glicuronidação no fígado. Um estudo da farmacocinética do raltegravir foi realizado em pacientes adultos com insuficiência hepática moderada. Além disso, a insuficiência hepática foi avaliada na análise farmacocinética composta.
Não houve diferença farmacocinética clinicamente importante entre os pacientes com insuficiência hepática moderada e os indivíduos saudáveis. Não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve a moderada. O efeito da insuficiência hepática grave sobre a farmacocinética do raltegravir não foi estudado.
Insuficiência Renal:
O clearance renal do fármaco inalterado é uma via de eliminação de menor importância. Um estudo da farmacocinética do raltegravir foi realizado em pacientes adultos com insuficiência renal grave. Além disso, a insuficiência renal foi avaliada na análise farmacocinética composta.
Não houve diferenças farmacocinéticas clinicamente compostas entre os pacientes com insuficiência renal grave e os indivíduos saudáveis. Não é necessário nenhum ajuste de dose. Como o grau em que o Raltegravir pode ser dialisável é desconhecido, deve-se evitar a administração prévia de uma sessão de diálise.
Polimorfismo da UGT1A1:
Não há evidências de que os polimorfismos comuns da UGT1A1 alterem a farmacocinética do raltegravir em uma extensão clinicamente significativa. Em uma comparação de 30 indivíduos adultos com genótipo *28/*28 (associado à atividade reduzida da UGT1A1) com 27 indivíduos adultos com genótipo tipo selvagem, a razão média geométrica (IC 90%) da AUC foi de 1,41 (0,96, 2,09).
Farmacodinâmica
Microbiologia:
O raltegravir em concentrações de 31 20 nM resultou em inibição de 95% (CI95) da disseminação viral (em relação a uma cultura não tratada infectada por vírus) em culturas de células linfóides T humanas infectadas com a linhagem de célula adaptada HIV-1 variante H9IIIB.
Além disso, o raltegravir em concentrações de 6 a 50 nM resultou em inibição de 95% da disseminação viral em culturas de células mononucleares de sangue periférico humano ativadas por mitógeno infectadas por isolados clínicos primários diversos de HIV-1, incluindo isolados de 5 subtipos não-B, e isolados resistentes a inibidores da transcriptase reversa e a inibidores da protease.
Em um ensaio de infecção de ciclo único, o raltegravir inibiu a infecção de 23 isolados de HIV representando 5 subtipos não-B e 5 formas recombinantes circulantes com valores CI50 variando de 5 a 12 nM. O raltegravir também inibiu a replicação de um isolado de HIV-2 quando avaliado em células CEMx174 (CI95= 6 nM).
Foi observada atividade antirretroviral aditiva a sinérgica quando células linfóides T humanas infectadas com H9IIIB variante do HIV-1 foram incubadas com o raltegravir em combinação com inibidores nucleosídeos análogos da transcriptase reversa (zidovudina, zalcitabina, estavudina, abacavir, tenofovir, didanosina ou lamivudina); inibidores não nucleosídeos da transcriptase reversa (efavirenz, nevirapina ou delavirdina); inibidores da protease (indinavir, saquinavir, ritonavir, amprenavir, lopinavir, nelfinavir ou atazanavir); ou com o inibidor de entrada do HIV em células (enfuvirtida).
Resistência ao Medicamento:
As mutações observadas na HIV-1 integrase que contribuíram para a resistência ao raltegravir (desenvolvida tanto in vitro como em pacientes tratados com o raltegravir) geralmente incluíram substituição tanto em Q148 (alterado para H, K, ou R) como em N155 (alterado para H) acrescida de uma ou mais mutações adicionais (por exemplo, L74I/M, E92Q, E138A/K, G140A/S, ou V151I). A substituição aminoácida no Y143C/H/R é outra via de resistência ao raltegravir.
Os vírus recombinantes com uma única mutação primária (Q148H, K ou R, ou N155H) apresentaram capacidade de replicação e sensibilidade reduzidas ao raltegravir in vitro. As mutações secundárias diminuem ainda mais a sensibilidade ao raltegravir e algumas vezes agiram como mutações compensatórias para capacidade de replicação viral.
Eletrofisiologia Cardíaca:
Em um estudo randômico, controlado com placebo e cruzado, 31 indivíduos saudáveis receberam uma dose única oral supraterapêutica de 1.600 mg de raltegravir e placebo. Não houve efeito sobre o intervalo QTc. Os picos das concentrações plasmáticas de raltegravir foram aproximadamente 4 vezes mais altos dos que os picos das concentrações após uma dose de 400 mg.
DCB (Denominação Comum Brasileira)
Nomes comerciais
Especialidades Médicas
Infectologia
Quer saber mais?
Consulta também a Bula do Raltegravir
Ler a bula do Raltegravir completa