Bula do Nexavar
Princípio Ativo:Tosilato de Sorafenibe
Classe Terapêutica:Outros Antineoplásicos Inibidores da Proteína Kinase
Nexavar, para o que é indicado e para o que serve?
Nexavar® é indicado para o tratamento de:
Um determinado tipo de câncer nos rins que não tenha respondido ao tratamento prévio com alfainterferona ou interleucina-2 ou que não pudessem receber tal terapia.
Tratamento de câncer no fígado que não possa ser removido com cirurgia.
Tratamento de pacientes com um tipo de câncer de tireoide (diferenciado - papilífero, folicular, célula de Hurthle) localmente avançado ou metastático, progressivo, que não responde à terapia com iodo radioativo.
Nexavar® deve ser prescrito e controlado por um oncologista ou profissional de saúde capacitado para tratar pacientes com câncer de rim, fígado ou tireoide.
Como o Nexavar funciona?
Nexavar® tem como substância ativa o sorafenibe, um inibidor de múltiplas quinases que atua diminuindo o crescimento das células cancerígenas e interrompendo o suprimento de sangue que mantém o crescimento dessas células.
As concentrações plasmáticas do sorafenibe no estado de equilíbrio são alcançadas em 7 dias, com uma relação máximo-mínimo das concentrações médias inferior a 2. Converse com o seu médico para que ele possa lhe dar mais detalhes sobre a ação deste medicamento.
Quais as contraindicações do Nexavar?
Nexavar® não deve ser usado se você tiver hipersensibilidade grave (alergia) ao sorafenibe ou a qualquer outro componente deste medicamento.
Como usar o Nexavar?
Uso oral.
A dose recomendada é de 2 comprimidos de Nexavar®, duas vezes ao dia, que corrresponde a 800 mg por dia.
Nexavar® deve ser engolido inteiro, com o auxílio de um pouco de água.
Pode-se tomá-lo entre as refeições ou durante refeições com pouca ou moderada quantidade de gordura. Não tome Nexavar® com refeições ricas em gordura, isto pode deixá-lo menos efetivo. Se você pretende ter uma refeição rica em gordura, Nexavar® deve ser tomado pelo menos 1 hora antes ou 2 horas após a refeição.
É importante tomar Nexavar® aproximadamente no mesmo horário todos os dias, assim haverá uma quantidade estável do medicamento na circulação sanguínea.
Duração do tratamento e ajuste de dose
O seu tratamento durará enquanto for benéfico.
Seu médico pode interromper temporariamente o tratamento ou modificar a quantidade de comprimidos ingeridos por dia, dependendo de você estar tolerando ou não o tratamento.
Informações adicionais para populações especiais
Pacientes idosos (acima de 65 anos), gênero e peso corporal
Nenhum ajuste de dose é necessário com base na idade (acima de 65 anos), no gênero ou no peso corporal do paciente.
Uso em crianças
A segurança e a eficácia de Nexavar® em pacientes pediátricos não foram estabelecidas.
Pacientes com insuficiência hepática
Não há necessidade de ajuste de dose em pacientes com insuficiência hepática Child-Pugh A ou B. Não se estudou Nexavar® em pacientes com insuficiência hepática Child-Pugh C.
Pacientes com insuficiência renal
Não há necessidade de ajuste de dose em pacientes com insuficiência renal leve, moderada ou grave que não requeira diálise. Nexavar® não foi estudado em pacientes que estejam sendo submetidos à diálise.
É recomendável que o médico monitore o equilíbrio hidroeletrolítico nos pacientes com risco de disfunção renal.
Este medicamento não deve ser partido, aberto ou mastigado.
Siga a orientação do seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Superdose: o que acontece se tomar uma dose do Nexavar maior do que a recomendada?
Não existe tratamento específico em caso de superdose de Nexavar®. A dose mais alta de Nexavar® estudada clinicamente foi de 4 comprimidos (800 mg) duas vezes ao dia. A ingestão de uma quantidade maior do que a indicada de Nexavar® pode tornar os efeitos adversos mais prováveis ou mais graves, especialmente diarreia e reações dermatológicas. Na suspeita de superdose, deve-se interromper o uso de Nexavar® e consultar seu médico imediatamente.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
O que devo fazer quando me esquecer de usar o Nexavar?
Se você esquecer de tomar uma dose de Nexavar®, deve tomá-la assim que se lembrar; mas se for perto do horário da próxima dose, você não deve ingerir a dose esquecida. Você não deve ingerir duas doses para compensar a dose esquecida.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Quais cuidados devo ter ao usar o Nexavar?
Informe seu médico se qualquer das situações a seguir se aplica a você. Você pode precisar de algum tratamento ou seu médico pode decidir alterar a dose de Nexavar® ou ainda suspender o tratamento com Nexavar®.
Se você apresentar reações cutâneas:
Nexavar® pode causar reações na pele, principalmente nas mãos e nos pés, e o rash cutâneo. Em geral, essas reações surgem durante as primeiras 6 semanas de tratamento com sorafenibe. O médico pode controlar as reações na pele com tratamento tópico para alívio dos sintomas, interromper o tratamento temporariamente e/ou modificar a dose do medicamento, ou, em casos graves ou persistantes, interromper o tratamento permanentemente.
Se você tem hipertensão (pressão alta):
Observou-se aumento da incidência de hipertensão (pressão alta) em pacientes que receberam Nexavar®. Em geral, a hipertensão foi leve a moderada, surgiu no início do tratamento e foi controlável com medicamentos habituais para pressão alta. O médico deve controlar regularmente a pressão arterial, e se necessário, iniciar tratamento de acordo com as condutas médicas padronizadas. Em caso de hipertensão grave, persistente ou de crises hipertensivas apesar do tratamento anti-hipertensivo adequado, o médico deve considerar a descontinuação permanente do tratamento com Nexavar®.
Se você apresenta sangramentos ou se está tomando varfarina (um anticoagulante):
O tratamento com Nexavar® pode aumentar o risco de hemorragia. A incidência de eventos hemorrágicos graves é incomum. Se ocorrer um sangramento que requeira atenção médica, poderá ser necessária a descontinuação do tratamento com Nexavar®. Em pacientes com câncer de tiroide diferenciado, no caso de haver infiltração da traqueia, dos brônquios e do esôfago deve ser feita terapia localizada antes de administrar Nexavar®, devido ao risco potencial de sangramento. Informe seu médico se estiver tomando varfarina, pois estes medicamentos alteram a consistência do sangue para prevenir formação de coágulo e podem aumentar o risco de hemorragia.
Se você apresentar dores no peito e problemas cardíacos:
Caso você desenvolva isquemia cardíaca e/ou infarto durante o tratamento com Nexavar®, o médico deve considerar a descontinuação temporária ou permanente do tratamento.
Se você tem uma anormalidade do coração conhecida como intervalo QT prolongado:
Nexavar® pode afetar o rítmo cardíaco. Caso você tenha síndrome congênita do QT longo, esteja em tratamento com altas doses cumulativas de antraciclina (outro medicamento para tratamento de câncer), esteja tomando certos medicamentos antiarrítmicos ou outros medicamentos que levem ao prolongamento do QT ou apresente distúrbios eletrolíticos como baixa concentração de potássio (hipocalemia), cálcio (hipocalcemia) ou magnésio (hipomagnesemia) no sangue, informe ao seu médico pois ele pode achar necessário solicitar exames de sangue e eletrocardiogramas, periodicamente, durante o tratamento com Nexavar®.
Se você for submeter-se ou submeteu-se à cirurgia recentemente:
Nexavar® pode afetar a cicatrização de feridas, então, caso necessite passar por uma cirurgia, informe ao profissional de saúde que você está tomando Nexavar®. Seu médico poderá pedir para você interromper temporariamente o tratamento com Nexavar® e decidirá quando reiniciá-lo.
Se você estiver tomando irinotecano ou docetaxel (que também são medicamentos para tratamento de câncer):
Informe seu médico sobre todos os medicamentos que estiver tomando e quando for tomar algum novo medicamento. Recomenda-se precaução na administração de Nexavar® junto com docetaxel ou compostos metabolizados/eliminados pela mesma via (UGT1A1) que Nexavar®, por exemplo, irinotecano pois Nexavar® pode aumentar os efeitos e, em particular, os efeitos colaterais destes medicamentos.
Se você estiver tomando neomicina (um antibiótico):
A administração de Nexavar® junto com neomicina pode diminuir o efeito de Nexavar®.
Se você tiver alteração grave do funcionamento do fígado:
Informe seu médico se estiver com a função do fígado alterada. Você pode apresentar efeitos colaterais mais graves quando utilizar Nexavar®.
Se você apresentar perfuração intestinal:
Casos de perfuração intestinal são incomuns. Em alguns casos não estava relacionada a um tumor intrabdominal evidente. O médico deverá interromper o tratamento com Nexavar®.
Se você tiver câncer da tireoide:
Seu médico irá monitorar os níveis sanguíneos de cálcio e de hormônios da tireoide.
Gravidez e lactação
Deve-se evitar a gravidez durante o tratamento com Nexavar®. Informe imediatamente seu médico em caso de suspeita de gravidez, pois este medicamento pode causar danos ao feto, que incluem malformação grave, desenvolvimento insuficiente e morte. Deve-se utilizar medidas eficazes para prevenir a gravidez durante o tratamento e por pelo menos 2 semanas após o término do tratamento.
Nexavar® não deve ser usado durante a gravidez. O médico irá considerar o uso deste medicamento por mulheres grávidas quando os possíveis benefícios justificarem os possíveis riscos para o feto.
A amamentação deve ser interrompida durante o tratamento com Nexavar®.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Os resultados de estudos em animais indicam que sorafenibe pode prejudicar a fertilidade masculina e feminina.
Dirigir veículos e operar máquinas
Não foram conduzidos estudos sobre o efeito de Nexavar® na habilidade de dirigir veículos ou de operar máquinas.
Quais as reações adversas e os efeitos colaterais do Nexavar?
Como todo medicamento, Nexavar® pode causar reações adversas, embora nem todos pacientes apresentem essas reações.
As reações adversas mais comuns foram diarreia, fadiga, queda de cabelo, infecção, reação cutânea mão-pé (desenvolvimento de vermelhidão, dor, inchaço, podendo evoluir com formação de bolhas, nas palmas das mãos ou nas solas dos pés) e vermelhidão/descamação da pele (rash).
Os eventos adversos mais comuns, que foram considerados relacionados ao Nexavar®, nos pacientes com câncer renal, hepático ou de tireoide foram fadiga, perda de peso, reação cutânea mão-pé, queda de cabelo, diarreia, falta de apetite, náusea, dor abdominal, hipertensão, infecção e sangramento.
As reações adversas observadas em estudo clínico para tratamento de câncer do fígado, relatadas em pelo menos 10% dos pacientes, em ordem decrescente de frequência, foram:
Diarreia, fadiga, dor abdominal, perda de peso, falta de apetite, náusea, reação cutânea mão-pé, vermelhidão/descamação da pele, vômitos, constipação, queda de cabelo, coceira, distúrbios do fígado e pele seca.
As reações adversas observadas em estudo clínico para tratamento de câncer renal, relatadas em pelo menos 10% dos pacientes, em ordem decrescente de frequência, foram:
Diarreia, vermelhidão/descamação da pele, fadiga, reação cutânea mão-pé, queda de cabelo, náusea, coceira, hipertensão, falta de apetite, vômito, constipação, hemorragia, falta de ar, distúrbios da sensibilidade da pele, pele seca, dor abdominal, perda de peso, dor nas articulações e dor de cabeça.
As reações adversas observadas em estudo clínico para tratamento de câncer de tireoide diferenciado, relatadas em pelo menos 10% dos pacientes, em ordem decrescente de frequência, foram:
Reação cutânea mão-pé, diarreia, queda de cabelo, vermelhidão/descamação da pele, fadiga, perda de peso, hipertensão, infecção, falta de apetite, inflamação da mucosa da cavidade oral (mucosite), náuseas, coceira, níveis baixos de cálcio no sangue, dor de cabeça, constipação, dor nas extremidades dos membros, pele seca, dor abdominal, distúrbios da sensibilidade da pele, elevações de enzimas produziadas pelo fígado, como ALT (alanina aminotrasferase) e AST (aspartato aminotransferase), alterações na voz, dor, vômito, febre e dor de garganta/faringe/laringe.
Reações adversas de acordo com a frequência
Reações adversas muito comuns (10% ou mais)
- Sensação de fraqueza ou cansaço;
- Dor (incluindo dor na cavidade oral, nos ossos e tumoral);
- Vermelhidão;
- Infecção;
- Hemorragia (incluindo hemorragia no intestino*, no trato respiratório* e casos incomuns de hemorragia cerebral*);
- Febre;
- Diminuição do apetite;
- Dor nas articulações (artralgia);
- Níveis baixos de fosfato no sangue;
- Baixa contagem de leucócitos e linfócitos (tipos de células brancas) no sangue;
- Níveis aumentados de amilase e lipase no sangue (exames relacionados ao pâncreas).
Reações adversas comuns (1 a < 10%)
- Quadro clínico semelhante ao da gripe;
- Má digestão (dispepsia);
- Inflamação da mucosa do trato gastrintestinal (mucosite);
- Dificuldade para engolir (disfagia);
- Dor/queimação na língua e cavidade oral, com lesões dolorosas, incluindo boca seca e inflamada (estomatite);
- Refluxo gastroesofágico;
- Dor nos músculos (mialgia);
- Espasmos musculares;
- Depressão;
- Alteração do paladar (disgeusia);
- Problemas de ereção (impotência);
- Alteração na voz (disfonia);
- Secreção nasal persistente (rinorreia);
- Acne;
- Vermelhidão na pele (rubor facial e do pescoço);
- Pele inflamada, seca ou escamosa podendo sangrar (dermatite esfoliativa);
- Infecção dos folículos pilosos (foliculite);
- Crescimento celular localizado benigno da pele (ceratoacantomas);
- Câncer de pele (carcinoma espinocelular);
- Espessamento da camada externa da pele (hiperqueratose);
- Alteração da função cardíaca, do tipo insuficiência cardíaca congestiva*;
- Isquemia miocárdica e/ou infarto do miocárdio*;
- Alteração da função dos rins;
- Baixa contagem de células sanguíneas (células brancas, vermelhas (anemia) ou plaquetas);
- Aumento temporário de enzimas produzidas pelo fígado, detectado em exames de sangue;
- Níveis baixos de cálcio no sangue (hipocalcemia);
- Níveis baixos de potássio no sangue (hipocalemia);
- Níveis baixos de sódio no sangue (hiponatremia);
- Glândula tireoide com atividade reduzida (hipotireoidismo);
- Níveis elevados de proteína na urina (proteinúria).
Reações adversas incomuns (0,1 a < 1%)
- Inflamação do revestimento do estômago (gastrite);
- Inflamação do pâncreas (pancreatite);
- Inflamação da vesícula biliar (colecistite) e/ou ductos biliares;
- Amarelamento da pele ou olhos (icterícia) causada por altos níveis de pigmentos da bile no sangue (hiperbilirrubinemia);
- Reações alérgicas (incluindo reações de pele e urticária);
- Reação anafilática (reação alérgica grave);
- Desidratação;
- Aumento do tamanho das mamas (ginecomastia);
- Eventos do tipo doença pulmonar intersticial (inflamação nos pulmões);
- Eczema;
- Reações da pele e/ou das membranas mucosas, graves, que podem incluir bolhas dolorosas (eritema multiforme);
- Glândula tireoide com atividade anormal aumentada (hipertireoidismo);
- Crises hipertensivas* (pressão arterial exageradamente alta);
- Perfuração gastrintestinal*;
- Edema reversível na parte posterior do cérebro que pode estar associado à dor de cabeça, alteração da consciência, convulsões e sintomas visuais incluindo perda da visão (leucoencefalopatia posterior reversível*);
- Aumento temporário dos níveis de fosfatase alcalina no sangue;
- Alteração na RNI (exame que avalia a coagulação do sangue);
- Zumbido.
Reações adversas raras (0,01 a < 0,1%)
- Ritmo cardíaco anormal (prolongamento do QT);
- Lesão renal com perda de grande quantidade de proteína na urina (síndrome nefrótica);
- Hepatite induzida por medicamentos* (inflamação do fígado, que pode causar náuseas, vômitos, dor abdominal e icterícia).
Outros efeitos adversos, observados na experiência pós-comercialização
A frequência desses eventos não pode ser estimada a partir dos dados disponíveis.
- Dermatite de “recall” induzida por radiação (lesão cutânea que pode ocorrer em local exposto previamente a radioterapia. Pode ser um evento grave);
- Síndrome de Stevens-Johnson e necrólise epidérmica tóxica* (condição grave que apresenta-se como vermelhidão, erupção e bolhas dolorosas na pele e nas membranas mucosas, incluindo descolamento extenso da pele);
- Vasculite leucocitoclástica (inflamação de vasos da pele que pode causar manifestações cutâneas);
- Angioedema (reação alérgica com edema (p. ex., de face e língua) que pode causar dificuldade para respirar ou engolir);
- Rabdomiólise (destruição anormal de células musculares que pode levar a problemas renais);
- Eventos do tipo doença pulmonar intersticial (inflamação dos pulmões).
*Essas reações adversas podem ser potencialmente fatais ou fatais. São incomuns ou têm incidência mais baixa que incomum.
Adicionalmente, os seguintes eventos adversos significativos foram relatados de modo infrequente durante os estudos clínicos de Nexavar®:
Ataque isquêmico transitório, arritmia cardíaca (distúrbios do ritmo cardíaco) e tromboembolismo (formação de coágulos dentro dos vasos e seu desprendimento). Para esses eventos não se estabeleceu uma relação causal com Nexavar®.
Alterações de exames laboratoriais
Nexavar® pode alterar os resultados de exames laboratoriais, portanto quando for realizar algum tipo de exame de laboratório, informe que está tomando Nexavar®.
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
Interação medicamentosa: quais os efeitos de tomar Nexavar com outros remédios?
Alguns medicamentos podem interferir no efeito de Nexavar® ou ter seu efeito alterado por ele.
Informe seu médico se você está tomando algum dos medicamentos a seguir:
- Antibióticos rifampicina ou neomicina;
- Erva de São João, tratamento fitoterápico para depressão;
- Fenitoína, carbamazepina ou fenobarbital, tratamentos para epilepsia e outras condições;
- Dexametasona, corticosteroide utilizado para várias condições;
- Varfarina, anticoagulante utilizado para prevenção de coágulos sanguíneos;
- Doxorrubicina, capecitabina, docetaxel, paclitaxel, carboplatina e irinotecano, que são outros tratamentos de câncer;
- Digoxina, para o tratamento de problemas cardíacos.
Informe seu médico se você está tomando, ou tomou recentemente, os medicamentos listados acima ou qualquer outro medicamento, inclusive medicamentos sem prescrição médica.
Interações com alimentos
Recomenda-se que Nexavar® seja tomado entre as refeições ou durante refeições com pouca ou moderada quantidade de gordura. Não se deve tomá-lo durante refeições ricas em gordura. Se você pretende ter uma refeição rica em gordura, Nexavar® deve ser tomado pelo menos 1 hora antes ou 2 horas após a refeição.
Quando Nexavar® é administrado durante uma refeição com gordura moderada, a biodisponibilidade é similar à registrada em jejum. Com uma refeição rica em gordura, a biodisponibilidade do sorafenibe é reduzida em 29% em comparação com a administração em jejum.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para sua saúde.
Apresentações do Nexavar
Nexavar® é apresentado na forma de comprimidos revestidos com 200 mg de sorafenibe em embalagem com 60 comprimidos.
Uso oral.
Uso adulto.
Qual a ação da substância do Nexavar?
Resultados de eficácia
A segurança e eficácia de Tosilato de Sorafenibe foram estudadas em pacientes com carcinoma hepatocelular (CHC), em pacientes com carcinoma de células renais (CCR) avançado e em pacientes com carcinoma de tireoide diferenciado (CTD).
Carcinoma hepatocelular
Estudo 3 (estudo 100554)
Foi um estudo Fase III internacional, multicêntrico, randomizado, duplo-cego e controlado com placebo, conduzido em 602 pacientes com carcinoma hepatocelular. O objetivo primário do estudo foi a sobrevida global e o objetivo secundário foi o tempo até a progressão (TTP).
As características demográficas e da doença no período basal foram comparáveis entre os grupos de Tosilato de Sorafenibe e placebo com relação a idade, gênero, raça, “performance status”, etiologia (incluindo hepatite B, hepatite C e doença hepática alcoólica), estadio TNM (estadio I: < 1% vs. < 1%; estadio II: 10,4% vs. 8,3%; estadio III: 37,8% vs. 43,6%; estadio IV: 50,8% vs. 46,9%), ausência de invasão vascular macroscópica e de disseminação extra-hepática do tumor (30,1% vs. 30,0%), e estadio BCLC (estadio B: 18,1% vs. 16,8%; estadio C: 81,6% vs. 83,2%; estadio D: < 1% vs. 0%). A função hepática de acordo com o índice de Child-Pugh foi comparável entre os grupos de Tosilato de Sorafenibe e placebo (A: 95% vs. 98%; B: 5% vs. 2%). Somente um paciente com disfunção hepática Child-Pugh C recebeu a medicação do estudo. Tratamentos prévios incluíram procedimentos de ressecção cirúrgica (19,1% vs. 20,5%), terapias locorregionais (incluindo ablação por radiofrequência, injeção percutânea de etanol e quimioembolização transarterial; 38,8% vs. 40,6%), radioterapia (4,3% vs. 5,0%) e terapia sistêmica (3,0% vs. 5,0%).
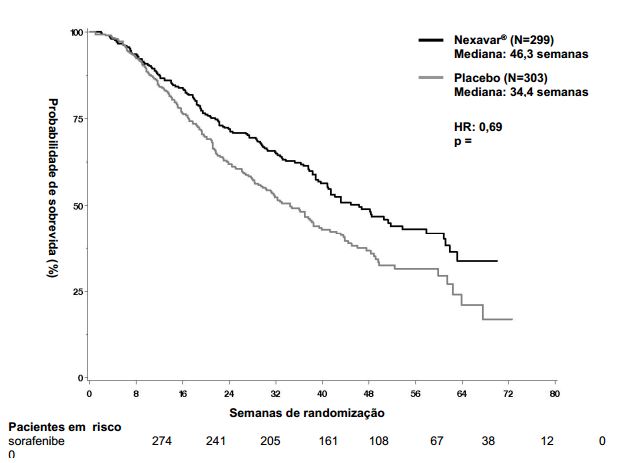
O estudo foi interrompido após uma análise interina planejada de sobrevida global haver ultrapassado o limite de eficácia pré-especificada. Esta análise mostrou uma vantagem estatisticamente significativa de Tosilato de Sorafenibe sobre o placebo para sobrevida global (HR: 0,69, p= 0,00058, veja tabela 1 e figura 1). Esta vantagem foi consistente em quase todos os subgrupos analisados. Nos fatores de estratificação pré-especificados (“performance status” por ECOG, presença ou ausência de invasão vascular macroscópica e/ou disseminação extra-hepática do tumor e região) o “hazard ratio” (HR) consistentemente favoreceu Tosilato de Sorafenibe sobre o placebo. O tempo até a progressão do tumor (TTP, por revisão radiológica independente) foi significativamente maior no grupo Tosilato de Sorafenibe (HR: 0,58, p=0,000007, veja tabela 1).
Tabela 1: Resultados de Eficácia do estudo 3 (estudo 100554) no carcinoma hepatocelular:
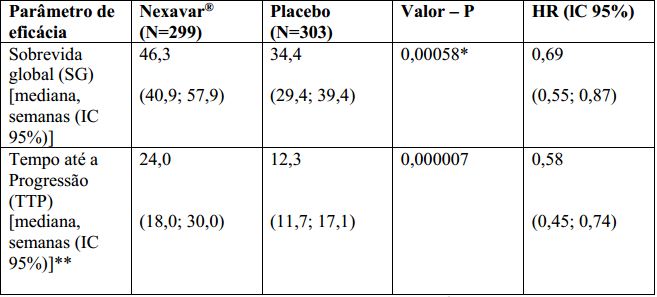
lC = intervalo de confiança, HR = “Hazard Ratio” (Tosilato de Sorafenibe sobre placebo).
*Estatisticamente significativa porque o valor de p foi inferior a 0,0077, limite de O’Brien Fleming pré-especificado.
** Revisão radiológica independente.
Figura 1: Curva de Kaplan-Meier de sobrevida global no estudo 3 (estudo 100554, população de “intenção de tratar”):
Carcinoma de células renais
A segurança e eficácia de Tosilato de Sorafenibe no tratamento de carcinoma de células renais avançado (CCR) foram estudadas nos dois estudos clínicos controlados randomizados a seguir.
Estudo 1 (11213)
Foi um estudo Fase III em 903 pacientes, internacional, multicêntrico, randomizado, duplo-cego e controlado com placebo. Os objetivos primários do estudo incluíram a sobrevida global e a sobrevida livre de progressão (SLP). A taxa de resposta tumoral foi um objetivo secundário.
Os pacientes foram randomizados para receber Tosilato de Sorafenibe 400 mg duas vezes ao dia (N = 451) ou placebo (N = 452). Ao início do estudo, as características demográficas e dos pacientes foram comparáveis em ambos os grupos de tratamento. Aproximadamente metade dos pacientes apresentou “performance status” por ECOG (“Eastern Cooperative Oncology Group”) de 0, e metade dos pacientes estava no grupo prognóstico de baixo risco do MSKCC (“Memorial Sloan Kettering Cancer Center”).
Numa análise interina de sobrevida, planejada ao início do estudo e baseada em 220 óbitos, houve uma melhora estimada de 39% da sobrevida global de pacientes que receberam sorafenibe em comparação com o placebo. A estimativa de HR (“hazard ratio” - risco de morte com sorafenibe comparado com placebo) foi de 0,72 (IC 95%, 0,55 - 0,95; p = 0,018). O limiar de significância estatística para a análise intermediária foi p < 0,0005). A probabilidade de sobrevida em 1 ano foi estimada em 64,9% para o sorafenibe e 57,7% para o placebo, resultando em um NNT (número necessário para tratar) de 13,9.
A análise da SLP incluiu 769 pacientes randomizados para receber Tosilato de Sorafenibe 400 mg duas vezes ao dia (N = 384) ou placebo (N = 385). A SLP foi avaliada por revisão radiológica independente mascarada usando critérios RECIST (“Response Evaluation Criteria in Solid Tumors”). A mediana de SLP foi o dobro para pacientes que receberam sorafenibe (167 dias) em comparação com os pacientes que receberam placebo (84 dias) (HR = 0,44; IC 95%: 0,35 - 0,55; p < 0,000001).
O efeito na SLP também foi explorado em vários subgrupos diferentes de pacientes. Os subgrupos incluíram idade acima ou abaixo de 65 anos, “performance status” por ECOG de 0 ou 1, categoria 1 do índice prognóstico do MSKCC, terapia prévia para doença metastática progressiva ou para doença em estadio mais precoce e tempo a partir do diagnóstico menor ou maior que 1,5 ano. O efeito de sorafenibe sobre a SLP foi consistente nestes subgrupos, inclusive para os pacientes que não receberam terapia anterior com IL-2 ou interferona (N = 137; 65 pacientes recebendo sorafenibe, 72 recebendo placebo), para os quais a SLP mediana foi de 172 dias para o sorafenibe, em comparação com 85 dias para o placebo (HR= 0,35; IC 95%: 0,19 - 0,63).
A melhor resposta tumoral global foi determinada por revisão radiológica feita pelo pesquisador de acordo com critérios RECIST. No grupo de sorafenibe, 1 paciente (0,2%) apresentou resposta completa, 43 pacientes (9,5%) apresentaram resposta parcial e 333 pacientes (73,8%) apresentaram doença estável. No grupo placebo, nenhum paciente (0%) apresentou resposta completa, 8 pacientes (1,8%) apresentaram resposta parcial e 239 pacientes (52,9%) apresentaram doença estável.
O tratamento com sorafenibe não resultou em deterioração geral dos sintomas específicos de câncer renal (FKSI-10) ou na qualidade de vida relacionada à saúde, em comparação com o placebo. Após 18 e 24 semanas de tratamento, mais pacientes que recebiam sorafenibe reportaram melhora do escore FKSI-10 total (55 e 44%, respectivamente) e do escore de bem-estar físico (“FACT-G PWB = Functional Assessment of Cancer Therapy – General version – Physical Well Being”) (57 e 47%, respectivamente) em comparação com o placebo (FKSI-10, 33 e 21%; e FACT-G PWB, 37 e 21%, respectivamente).
O maior tempo de tratamento com sorafenibe verificado no estudo 1 foi de 72 semanas.
Estudo 2
Foi um estudo Fase II, de descontinuação randomizada, que incluiu pacientes com neoplasias malignas metastáticas, incluindo o carcinoma de células renais. O objetivo primário deste estudo foi a porcentagem de pacientes randomizados (N = 65), que permaneceram sem progressão após 24 semanas. Os pacientes randomizados para sorafenibe apresentaram SLP mediana significativamente mais prolongada (163 dias) que os pacientes randomizados para placebo (41 dias; valor de p = 0,0001; HR 0,29). A taxa de SLP foi significativamente mais alta no grupo de sorafenibe (50%) que no placebo (18%) (p = 0,0077).
O maior tempo de tratamento com sorafenibe verificado no estudo 2 foi de 155 semanas.
Carcinoma de Tireoide Diferenciado
O estudo 4 (14295) foi um estudo fase III, internacional, multicêntrico, randomizado, duplo cego, controlado com placebo, em 417 pacientes com carcinoma de tireoide diferenciado (CTD) localmente avançado ou metastático, refratário a iodo radioativo (RAI).
O desfecho primário do estudo foi a sobrevida livre de progressão (SLP). Os desfechos secundários incluíram a sobrevida global (SG), a taxa de resposta tumoral e a duração da resposta. Os pacientes que apresentaram progressão foram autorizados a receber Tosilato de Sorafenibe em esquema aberto. Não foi permitido tratamento concomitante com iodo radioativo.
O estudo de fase III (14295) recrutou pacientes com CTD histologicamente comprovado, localmente avançado ou metastático, refratário ao tratamento com iodo radioativo que tinham progredido de acordo com RECIST nos 14 meses anteriores ao recrutamento. CTD refratário a iodo radioativo foi definido como presença de uma lesão não captante de iodo em um scan com iodo radioativo, ou utilização de doses radioativas ≥ 600 mCi de iodo radioativo, ou progressão após tratamento com iodo radioativo dentro de 16 meses após o início do recrutamento ou após dois tratamentos com iodo radioativo separados por intervalos de 16 meses.
As características demográficas e dos pacientes no período basal foram bem balanceadas entre os dois grupos de tratamento. Oitenta e seis por cento (86%) dos pacientes apresentavam metástases nos pulmões, 51% nos linfonodos e 27% nos ossos. Quase todos os pacientes haviam sido submetidos à tireoidectomia (99,5%) e haviam recebido radioatividade cumulativa mediana de aproximadamente 400 mCi. A maioria dos pacientes tinha carcinoma papilífero (56,8%), seguido por carcinoma folicular (25,4%) e carcinoma pouco diferenciado (9,6%). Os pacientes incluídos no estudo apresentaram “performance status” ECOG 0,1,2 sendo que aproximadamente 60% dos pacientes apresentaram “performance status” ECOG 0.
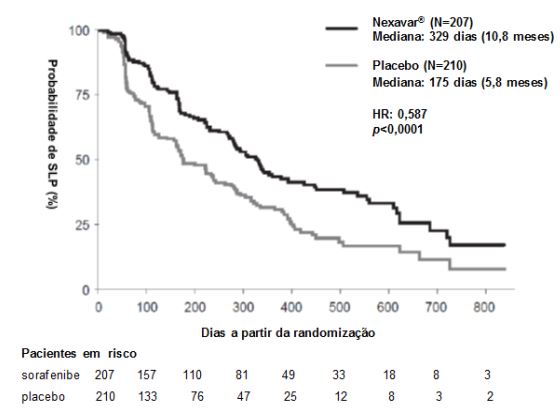
O conjunto completo para análise incluiu 207 pacientes randomizados para Tosilato de Sorafenibe 400 mg duas vezes ao dia e 210 pacientes randomizados para placebo. A SLP foi avaliada por revisão radiológica independente, cega, com base nos critérios RECIST. A mediana do tempo de SLP foi de 329 dias (10,8 meses) no grupo Tosilato de Sorafenibe comparada a 175 dias (5,8 meses) no grupo placebo. O risco relativo para a SLP (progressão da doença ou morte) foi reduzido em aproximadamente 41% nos pacientes que receberam sorafenibe em comparação com os indivíduos que receberam placebo (relação de risco [“Hazard Ratio” (HR)] = 0,587, Intervalo de confiança (IC) 95%: 0,454; 0,758; p unilateral <0,0001). (Tabela 2, Figura 2)
Não houve diferença estatística significamente na sobrevida global entre os grupos de tratamento (a HR foi 0,884, IC 95%: 0,633; 1,236, valor p unilateral de 0,236, Tabela 2) na análise da sobrevida global realizada 9 meses após a data de corte da análise final de SLP. A SG mediana não foi alcançada no braço sorafenibe e foi de 36,5 meses no braço placebo. Cento e cinquenta e sete (75%) pacientes randomizados para placebo e 61 (30%) randomizados para Tosilato de Sorafenibe receberam Tosilato de Sorafenibe em esquema aberto. A duração mediana da terapia no período duplo cego foi de 46 semanas (faixa 0,3 – 135) para pacientes recebendo sorafenibe e 28 semanas (faixa 1,7 – 132) para pacientes recebendo placebo. Não foi observada resposta completa (RC) de acordo com RECIST. A taxa de resposta global (RC + resposta parcial (RP)), por avaliação radiológica independente, foi mais alta no grupo de Tosilato de Sorafenibe (24 pacientes, 12,2%) do que no grupo placebo (um paciente, 0,5%), p unilateral <0,0001. A duração mediana da resposta foi de 309 dias (IC 95%: 226, 505 dias) entre os pacientes que receberam Tosilato de Sorafenibe e apresentaram RP.
Uma análise de subgrupo “post-hoc” sobre o tamanho máximo do tumor mostrou um efeito do tratamento para sobrevida livre de progressão favorecendo o sorafenibe com relação ao placebo para pacientes com tamanho máximo de tumor de 1,5 cm ou maior (HR 0,54; IC 95%: 0,41 – 0,71), enquanto um efeito numericamente menor foi relatado em pacientes com tamanho máximo de tumor menor que 1,5 cm (HR 0,87; IC 95%: 0,40 – 1,89).
Uma análise de subgrupo “post-hoc” sobre os sintomas iniciais (“baseline”) do carcinoma de tireoide mostrou um efeito do tratamento para sobrevida livre de progressão favorecendo o sorafenibe com relação ao placebo para pacientes sintomáticos e assintomáticos. O HR da sobrevida livre de progressão foi de 0,39 (IC 95%: 0,21 – 0,72) para pacientes com sintomas inciais (“baseline”) e de 0,60 (IC 95%: 0,45 – 0,81) para pacientes sem sintomas inciais (“baseline”).
Tabela 2: Resultados de eficácia do estudo 4 em carcinoma de tireoide diferenciado:
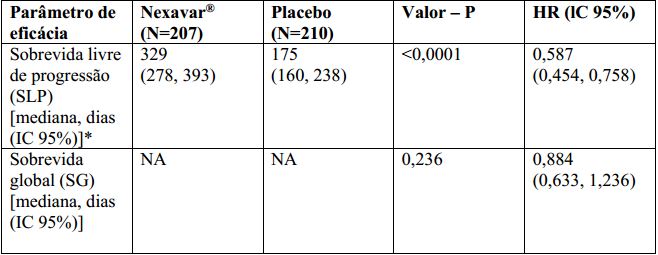 NA = Não alcançado IC = Intervalo de confiança, HR = “Hazard Ratio” (Tosilato de Sorafenibe sobre placebo).
NA = Não alcançado IC = Intervalo de confiança, HR = “Hazard Ratio” (Tosilato de Sorafenibe sobre placebo).
*Revisão radiológica independente.
Figura 2: Curva de Kaplan-Meier de sobrevida livre de progressão (SLP) em carcinoma de tireoide no estudo 4 (Conjunto completo para análise):
Características Farmacológicas
Farmacodinâmica
O sorafenibe é um inibidor de proteínas quinases.
Mecanismo de ação
O sorafenibe é um inibidor de múltiplas quinases, que reduz a proliferação celular tumoral in vitro.
O sorafenibe demonstrou inibir múltiplas quinases intracelulares (c-CRAF, BRAF e BRAF mutante) e da superfície celular (KIT, FLT-3, RET, RET/PTC, VEGFR-1, VEGFR-2, VEGFR-3 e PDGFR-beta). Acredita-se que várias dessas quinases estejam envolvidas na sinalização nas células tumorais, na angiogênese e na apoptose. O sorafenibe inibiu o crescimento tumoral de carcinoma hepatocelular, carcinoma de células renais e carcinoma de tireoide diferenciado humanos e de diversos outros xenoenxertos tumorais humanos em camundongos imunocomprometidos. Foram observados redução da angiogênese tumoral em modelos humanos de carcinomas hepatocelular e celular renal no tratamento com sorafenibe, e aumento da apoptose tumoral em modelos de carcinoma hepatocelular, carcinoma de células renais e carcinoma de tireoide diferenciado. Adicionalmente foi observada redução de sinalização na célula tumoral em modelos humanos de carcinoma hepatocelular e de carcinoma de tiroide diferenciado.
Prolongamento do intervalo QT
Em um estudo de farmacologia clínica, medidas de QT/QTc foram registradas em 31 pacientes no estado basal (pré-tratamento) e póstratamento. Após um ciclo de tratamento de 28 dias, no tempo de concentração máxima de sorafenibe, o QTcB foi prolongado em 4 ± 19 mseg e o QTcF em 9 ± 18 mseg, comparado ao tratamento placebo no estado basal. Nenhum paciente mostrou um QTcB ou QTcF > 500 mseg durante o monitoramento por ECG no pós-tratamento.
Farmacocinética
Absorção e distribuição
Após administração dos comprimidos de sorafenibe, a biodisponibilidade relativa média é de 38 – 49%, quando comparada com uma solução oral. Após administração oral, o sorafenibe atinge níveis plasmáticos máximos em aproximadamente 3 horas. Quando é administrado durante uma refeição com teores moderados de gordura, a biodisponibilidade é similar à registrada em jejum. Com uma refeição rica em gordura, a biodisponibilidade do sorafenibe é reduzida em 29% em comparação com a administração em jejum.
A ligação do sorafenibe às proteínas plasmáticas humanas in vitro é de 99,5%.
Metabolismo e biotransformação
O sorafenibe é metabolizado principalmente pelo fígado, sofrendo metabolismo oxidativo mediado pelo CYP3A4, assim como glicuronização mediada pelo UGT1A9.
O sorafenibe conjugado pode ser clivado no trato gastrintestinal pela atividade da glicuronidase bacteriana, o que permite a reabsorção do fármaco não conjugado. A coadministração de neomicina interfere neste processo, diminuindo a biodisponibilidade média do sorafenibe em 54%.
No estado de equilíbrio o sorafenibe representa 70-85%, aproximadamente, dos analitos circulantes no plasma. Foram identificados oito metabólitos do sorafenibe, dos quais cinco no plasma. O principal metabólito circulante no plasma, a N-óxido piridina, demonstrou potência in vitro semelhante à do sorafenibe e representa aproximadamente 9-16% dos analitos circulantes no estado de equilíbrio.
Eliminação/Excreção
Após administração oral de uma dose de 100 mg de uma formulação de solução de sorafenibe, 96% da dose foi recuperada em 14 dias: 77% excretada nas fezes e 19% excretada na urina, como metabólitos glicuronidados. Foram encontrados 51% da dose como sorafenibe intacto nas fezes, porém não na urina.
A meia-vida de eliminação do sorafenibe é de cerca de 25 – 48 horas.
Linearidade/Não-linearidade
Com doses superiores a 400 mg administradas duas vezes ao dia por via oral, as Cmáx e AUC médias aumentam abaixo da proporcionalidade esperada.
Farmacocinética no estado de equilíbrio
A administração de doses múltiplas de sorafenibe por 7 dias resultou em acúmulo 2,5 a 7 vezes superior comparada à administração de dose única.
As concentrações plasmáticas do sorafenibe no estado de equilíbrio são alcançadas em 7 dias, com uma relação vale/pico das concentrações médias inferior a 2.
A farmacocinética de sorafenibe no estado de equilíbrio na posologia de 400 mg duas vezes ao dia foi avaliada em pacientes com carcinoma de tireoide, CCR e CHC. A exposição média mais alta foi observada em pacientes com carcinoma de tireoide, embora a variabilidade na exposição tenha sido alta para todos os tipos de tumores. A relevância clínica do aumento da AUC em pacientes com carcinoma de tireoide é desconhecida.
Tabela 3: AUC(0-12)ss plasmática de sorafenibe no estado de equilíbrio de pacientes com carcinoma de tireoide diferenciado, CCR e CHC (média geométrica (%CV) [intervalo]):


Informações adicionais para populações especiais
Estudos sobre inibição enzimática
Os estudos realizados em microssomos hepáticos humanos demonstraram que sorafenibe é um inibidor competitivo de CYP2C19, CYP2D6 e CYP3A4. A administração clínica concomitante de midazolam, dextrometorfano e omeprazol, que são substratos dos citocromos CYP3A4, CYP2D6 e CYP2C19, respectivamente, após 4 semanas de administração do sorafenibe, não alterou a exposição à esses agentes, indicando que o sorafenibe não é indutor nem inibidor destas isoenzimas do citocromo P450.
Dados in vitro mostram que o sorafenibe inibe a glicuronização por meio das vias UGT1A1 (Ki=1µM) e UGT1A9 (Ki=2µM). A administração clínica concomitante de sorafenibe com irinotecano, cujo metabólito ativo SN-38 segue sendo metabolizado pela via UGT1A1, resultou em um aumento de 67 – 120% na AUC do SN-38. A exposição sistêmica a substratos de UGT1A1 e UGT1A9 pode aumentar quando coadministrados com o sorafenibe.
O sorafenibe inibe CYP2B6 e CYP2C8 in vitro, com valores de Ki de 6 µM e 1 - 2 µM, respectivamente. A administração clínica concomitante de sorafenibe com paclitaxel resultou em um aumento, ao invés de diminuição, na exposição ao paclitaxel 6-OH, metabólito ativo do paclitaxel que é formado pelo CYP2C8. Esses dados sugerem que sorafenibe pode não ser um inibidor in vivo do CYP2C8.
A administração concomitante de sorafenibe e ciclofosfamida resultou em uma pequena diminuição na exposição à ciclofosfamida, mas não diminuiu a exposição sistêmica à 4-OH ciclofosfamida, metabólito ativo da ciclofosfamida formado principalmente pelo CYP2B6. Esses dados sugerem que o sorafenibe pode não ser um inibidor in vivo do CYP2B6.
Os estudos realizados com microssomos hepáticos humanos demonstraram que sorafenibe é um inibidor competitivo de CYP2C9, com um valor de Ki = 7 – 8 µM. O possível efeito de sorafenibe sobre um substrato CYP2C9 foi avaliado em pacientes que receberam sorafenibe ou placebo em combinação com varfarina. Nos pacientes que receberam sorafenibe, as alterações médias de PT-RNI a partir dos valores basais não foram superiores àquelas dos pacientes que receberam placebo, sugerindo que sorafenibe pode não ser um inibidor de CYP2C9 in vivo.
Efeitos dos inibidores do CYP3A4
O cetoconazol (400 mg), um potente inibidor do CYP3A4, administrado uma vez ao dia durante 7 dias a voluntários sadios do sexo masculino, não alterou a AUC média de uma dose única de 50 mg de sorafenibe. Portanto, interações farmacocinéticas clínicas do sorafenibe com inibidores do CYP3A4 são improváveis.
Efeitos nos indutores do CYP
As atividades dos CYP1A2 e CYP3A4 não foram alteradas após tratamento de cultura de hepatócitos humanos com sorafenibe, indicando que é improvável que o sorafenibe seja um indutor do CYP1A2 e CYP3A4.
A administração clínica concomitante contínua de sorafenibe e rifampicina resultou em uma redução média de 37% da AUC do sorafenibe. Outros indutores da atividade do CYP3A4 (p. ex., Hypericum perforatum, também conhecida como Erva de São João, fenitoína, carbamazepina, fenobarbital e dexametasona) também podem aumentar o metabolismo do sorafenibe e, portanto, diminuir a sua concentração.
Combinação com outros agentes antineoplásicos
Em estudos clínicos, administrou-se sorafenibe em combinação com uma série de outros agentes antineoplásicos, em seus regimes de dose comumente utilizados, incluindo gencitabina, cisplatina, oxaliplatina, paclitaxel, carboplatina, capecitabina, doxorrubicina, docetaxel, irinotecano e ciclofosfamida. O sorafenibe não teve efeito clinicamente relevante na farmacocinética da gencitabina, cisplatina, carboplatina, oxaliplatina ou ciclofosfamida.
Paclitaxel / carboplatina
A administração de paclitaxel (225 mg/m2) e carboplatina (AUC = 6) com sorafenibe (≤ 400 mg duas vezes ao dia), administrados com uma pausa de 3 dias na administração de sorafenibe em torno da administração de paclitaxel / carboplatina, não resultou em efeito significante na farmacocinética do paclitaxel.
A coadministração de paclitaxel (225 mg/m2, uma vez a cada 3 semanas) e carboplatina (AUC = 6) com sorafenibe (400 mg duas vezes ao dia, sem pausa na administração de sorafenibe) resultou em um aumento de 47% da exposição ao sorafenibe, 29% da exposição ao paclitaxel e 50% da exposição ao 6-OH paclitaxel. A farmacocinética da carboplatina não foi afetada.
Esses dados indicam que não é necessário ajuste de dose quando paclitaxel e carboplatina são coadministrados com sorafenibe com pausa de 3 dias na administração de sorafenibe. A relevância clínica do aumento da exposição ao sorafenibe e ao paclitaxel, após a coadministração sem pausa na administração de sorafenibe é desconhecida.
Capecitabina
A coadministração de capecitabina (750 – 1.050 mg/m2, duas vezes ao dia, dias 1 – 14 a cada 21 dias) e sorafenibe (200 ou 400 mg duas vezes ao dia, administração contínua ininterrupta) não resultou em alteração significativa da exposição ao sorafenibe, mas em um aumento de 15 – 50% da exposição à capecitabina e em um aumento de 0 – 52% da exposição à 5-FU. A relevância clínica deste aumento leve a moderado na exposição à capecitabina e ao 5-FU quando coadministrados com sorafenibe é desconhecida.
Doxorrubicina / irinotecano
O tratamento concomitante com sorafenibe resultou em aumento de 21% na AUC da doxorrubicina.
Quando se administrou com irinotecano, cujo metabólito ativo SN-38 segue sendo metabolizado pela via UGT1A1, produziu-se aumento de 67 – 120% da AUC do SN-38 e aumento de 26 – 42% da AUC do irinotecano. Não se conhece a relevância clínica desses resultados.
Docetaxel
A coadministração de sorafenibe (200 mg duas vezes/dia ou 400 mg duas vezes/dia, administrados do 2° ao 19° dias de cada ciclo de 21 dias) e docetaxel (75 ou 100 mg/m², administrado uma vez a cada 21 dias) com uma pausa de 3 dias em torno da administração de docetaxel resultou em um aumento de 36–80% na AUC de docetaxel e de 16–32% na Cmáx de docetaxel. Recomenda-se cautela ao administrar sorafenibe com docetaxel.
Combinação com antibióticos
Neomicina
A coadministração de neomicina, um agente antimicrobiano não sistêmico, utilizado na erradicação da flora gastrintestinal, interfere com a reciclagem entêrohepática do sorafenibe resultando na diminuição da exposição a esse fármaco. Em voluntários sadios, tratados com neomicina por 5 dias, a biodisponibilidade média do sorafenibe diminuiu em 54%. A relevância clínica desses achados é desconhecida. Efeitos de outros antibióticos não foram estudados, mas provavelmente dependerão da capacidade do antibiótico em diminuir a atividade da glicuronidase.
Combinação com inibidores da bomba de prótons
Omeprazol
A coadministração de omeprazol não tem impacto na farmacocinética do sorafenibe. Não é necessário ajustar a dose do sorafenibe.
Pacientes pediátricos
Não se dispõem de dados farmacocinéticos em pacientes pediátricos.
Pacientes geriátricos
A análise dos dados demográficos sugere que não é necessário efetuar ajustes de dose por idade.
Pacientes com insuficiência hepática
O sorafenibe é depurado principalmente pelo fígado.
Em pacientes com carcinoma hepatocelular com insuficiência hepática leve (Child-Pugh A) ou moderada (Child-Pugh B), os valores de exposição ficaram dentro do intervalo observado em pacientes sem insuficiência hepática. A farmacocinética do sorafenibe em pacientes sem carcinoma hepatocelular, mas que apresentavam insuficiência hepática leve (Child-Pugh A) ou moderada (Child-Pugh B), foi similar à farmacocinética de voluntários sadios. A farmacocinética de sorafenibe não foi estudada em pacientes com insuficiência hepática grave (Child-Pugh C).
Pacientes com insuficiência renal
Em um estudo clínico farmacológico, a farmacocinética do sorafenibe foi avaliada após administração de uma dose única de 400 mg a indivíduos com função renal normal e a indivíduos com insuficiência renal leve (ClCr 50 – 80 mL/min), moderada (ClCr 30 a < 50 mL/min) ou insuficiência renal grave (ClCr < 30 mL/min) sem necessidade de diálise.
Não foi observada nenhuma relação entre a exposição ao sorafenibe e a função renal. Não é necessário efetuar ajustes de dose na insuficiência renal leve, moderada ou grave que não requer diálise.
Gênero
A análise dos dados demográficos sugere que não é necessário efetuar ajustes de dose por gênero.
Dados de segurança pré-clínicos
O perfil de segurança pré-clínico de sorafenibe foi avaliado em camundongos, ratos, cães e coelhos.
Toxicidade sistêmica
Estudos de toxicidade com doses repetidas revelaram alterações leves a moderadas (degenerações e regenerações) em vários órgãos.
Após administração repetida a cães jovens e em crescimento, foram observados efeitos nos ossos e dentes. As alterações consistiram de espessamento irregular das epífises femurais, com uma dose diária de sorafenibe de 600 mg/m2 de superfície corporal (equivalente a 1,2 vezes a dose terapêutica recomendada de 500 mg/m2 de superfície corporal); hipocelularidade da medula óssea próxima das epífises alteradas, com a dose de 200 mg/m2/dia; e alterações da composição da dentina, com 600 mg/m2/dia.
Não foram induzidos efeitos semelhantes em cães adultos.
Efeitos genotóxicos positivos foram obtidos para o sorafenibe em um ensaio in vitro de clastogenicidade (aberrações cromossômicas) em células de mamíferos (ovário de hamster chinês) na presença de ativação metabólica. Uma substância intermediária no processo de fabricação, que também está presente no fármaco final (< 0,15%), foi positiva para mutagênese em um ensaio bacteriano in vitro (teste de Ames). O sorafenibe não foi genotóxico no teste de Ames (o material continha 0,34% da substância intermediária) e em um ensaio in vivo com micronúcleos de camundongo.
Toxicidade reprodutiva
O sorafenibe mostrou-se embriotóxico e teratogênico quando administrado a ratos e coelhos. Os efeitos observados incluíram reduções nos pesos corporais materno e fetal, aumento do número de reabsorções fetais e aumento do número de malformações externas e viscerais. Os desfechos adversos para o feto foram observados com uma dose oral de 6 mg/m2/dia em ratos e 36 mg/m2/dia em coelhos.
Genotoxicidade e carcinogenicidade
Não foram realizados estudos de carcinogenicidade com sorafenibe.
Não foram realizados estudos específicos com sorafenibe em animais para avaliar o efeito na fertilidade. Entretanto, pode-se esperar um efeito adverso sobre a fertilidade masculina e feminina, pois os estudos com doses repetidas em animais mostraram alterações nos órgãos reprodutores de machos e fêmeas.
Qual a composição do Nexavar?
Cada comprimido de Nexavar® contém:
274 mg de tosilato de sorafenibe correspondente a 200 mg de sorafenibe.
Excipientes: croscarmelose sódica, celulose microcristalina, hipromelose, laurilsulfato de sódio, estearato de magnésio, macrogol, dióxido de titânio e óxido de ferro vermelho.
Como devo armazenar o Nexavar?
Nexavar® deve ser guardado na sua embalagem original protegido da umidade e à temperatura ambiente (entre 15°C e 30°C).
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características organolépticas
Nexavar® apresenta-se na forma de comprimido redondo de cor vermelha, sem cheiro característico.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais do Nexavar
MS-1.7056.0029
Farm. Resp.:
Dra. Dirce Eiko Mimura
CRF-SP n° 16532
Fabricado por:
Bayer AG
Leverkusen - Alemanha
Importado por:
Bayer S.A.
Rua Domingos Jorge, 1.100
04779-900 - Socorro – São Paulo - SP
C.N.P.J. nº 18.459.628/0001-15
SAC
0800 7021241
sac@bayer.com
Venda sob prescrição médica.
Doenças relacionadas
Quer saber mais?
Consulta também aBula do Tosilato de Sorafenibe
O conteúdo desta bula foi extraído manualmente da bula original, sob supervisão técnica do(a) farmacêutica responsável: Karime Halmenschlager Sleiman (CRF- PR 39421). Última atualização: 18 de Março de 2025.