
Voriconazol Farma Vision Importadora e Exportadora de Medicamentos 200mg, caixa com 10 frascos-ampola com pó para solução de uso intravenoso
Selecione a variação do produto
Voriconazol Farma Vision Importadora e Exportadora de Medicamentos 200mg, caixa com 10 frascos-ampola com pó para solução de uso intravenoso
- Voriconazol
Branca Comum (Venda Sob Prescrição Médica)
Temperatura ambiente
Não pode ser partido
Bula do Voriconazol Farma Vision Importadora e Exportadora de Medicamentos
Voriconazol Farma Vision Importadora e Exportadora de Medicamentos, para o que é indicado e para o que serve?
Voriconazol é um agente antifúngico triazólico de amplo espectro, e é indicado para tratamento de aspergilose invasiva; tratamento de infecções invasivas graves por Candida, incluindo candidemia e candidíase esofágica (incluindo C. krusei) e; tratamento de infecções fúngicas graves causadas por Scedosporium spp. e Fusarium spp.
Voriconazol deve ser administrado principalmente a pacientes com infecções progressivas e passíveis de causar a morte.
Quais as contraindicações do Voriconazol Farma Vision Importadora e Exportadora de Medicamentos?
Voriconazol é contraindicado a pacientes com hipersensibilidade conhecida ao voriconazol ou a qualquer componente da fórmula.
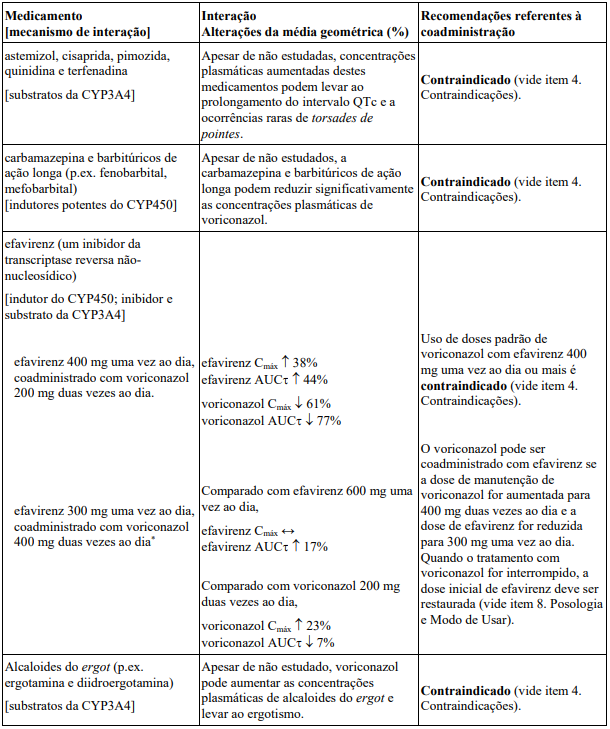
A coadministração de substratos do CYP3A4, tais como terfenadina, astemizol, cisaprida, pimozida ou quinidina com Voriconazol é contraindicada, uma vez que o aumento da concentração plasmática desses fármacos pode levar ao prolongamento do intervalo QTc e ocorrências raras de torsade de pointes.
A coadministração de Voriconazol e sirolimo está contraindicada uma vez que voriconazol pode causar aumento significativo das concentrações plasmáticas de sirolimo em indivíduos sadios.
A coadministração de Voriconazol com rifabutina, rifampicina, carbamazepina e barbitúricos de longa ação (ex.: fenobarbital) é contraindicada, uma vez que estes fármacos podem provocar decréscimo significativo das concentrações plasmáticas de voriconazol.
A coadministração de doses padrão de Voriconazol com doses de efavirenz de 400 mg uma vez ao dia ou superior é contraindicada, porque o efavirenz reduz significativamente a concentração plasmática de voriconazol em indivíduos saudáveis nestas doses. O voriconazol também aumenta significativamente a concentração plasmática de efavirenz.
A coadministração de Voriconazol e altas doses de ritonavir (400 mg e mais que duas vezes ao dia) está contraindicada uma vez que o ritonavir diminui significativamente a concentração plasmática de voriconazol nesta dose em indivíduos sadios.
A coadministração de alcaloides do ergot (ergotamina, diidroergotamina), os quais são substratos de CYP3A4, é contraindicada, uma vez que o aumento das concentrações plasmáticas desses fármacos pode levar ao ergotismo.
A coadministração de Voriconazol com Erva de São João é contraindicada.
Este medicamento é contraindicado para menores de 2 anos.
Como usar o Voriconazol Farma Vision Importadora e Exportadora de Medicamentos?
Comprimido
Voriconazol comprimidos revestidos deve ser administrado pelo menos uma hora antes ou uma hora após a refeição.
Uso em Adultos
A terapia com Voriconazol deve ser iniciada com o regime de dose de ataque intravenoso, para se obter no Dia 1, concentrações plasmáticas adequadas. O tratamento intravenoso deve continuar por pelo menos 7 dias antes da troca para a terapia oral. Uma vez que o paciente está clinicamente melhor e torna-se tolerante a medicação administrada por via oral, o comprimido de voriconazol pode ser utilizado. Devido à alta biodisponibilidade oral (96%), a troca entre a administração intravenosa e a oral é adequada, quando indicada clinicamente.
Informações detalhadas das recomendações de dosagem são apresentadas na tabela a seguir:
|
Dose de Manutençãoa |
||
|
Pacientes com 40 kg ou mais |
Pacientes com menos de 40 kg |
|
|
Aspergilose invasivab |
200 mg a cada 12 horas |
100 mg a cada 12 horas |
|
Infecções invasivas graves por Candida, inclusive candidemia |
200 mg a cada 12 horas |
100 mg a cada 12 horas |
|
Candidíase esofágica |
200 mg a cada 12 horas |
100 mg a cada 12 horas |
|
Scedosporioses e Fusarioses |
200 mg a cada 12 horas |
100 mg a cada 12 horas |
a. Em estudos com voluntários sadios, a dose oral de 200 mg a cada 12 horas resultou em uma exposição (AUCτ) similar à dose IV de 3 mg/kg a cada 12 horas, a dose oral de 300 mg a cada 12 horas resultou em uma exposição (AUCτ) similar à dose IV de 4 mg/kg a cada 12 horas.
b. A duração mediana da terapia oral de voriconazol foi de 76 dias (variação 2 – 232 dias).
Ajuste de Dose
Caso a resposta do paciente seja inadequada, a dose de manutenção deve ser aumentada de 200 mg a cada 12 horas (similar à dose IV de 3 mg/kg a cada 12 horas) para 300 mg a cada 12 horas (similar à dose IV de 4 mg/kg a cada 12 horas), para administração oral. Para os pacientes pesando menos de 40 kg, a dose oral de manutenção pode ser aumentada de 100 mg para 150 mg a cada 12 horas.
Se os pacientes não tolerarem o tratamento com altas doses (ex. 300 mg via oral a cada 12 horas), reduzir a dose oral de manutenção para intervalos de 50 mg até a dose mínima de 200 mg a cada 12 horas (ou 100 mg a cada 12 horas para pacientes adultos com peso inferior a 40 kg).
A fenitoína pode ser coadministrada com Voriconazol se a dose de manutenção oral for aumentada de 200 mg para 400 mg, a cada 12 horas por via oral (de 100 mg para 200 mg, a cada 12 horas, em pacientes com menos de 40 kg).
Quando Voriconazol é coadministrado com doses ajustadas de efavirenz, a dose de manutenção de Voriconazol deve ser aumentada para 400 mg a cada 12 horas.
A duração do tratamento depende da resposta clínica e micológica dos pacientes.
Uso em Pacientes Idosos
Não é necessário ajuste de dose em pacientes idosos.
Uso em Pacientes com Insuficiência Renal
A farmacocinética do voriconazol administrado por via oral não é afetada pela insuficiência renal. Portanto, não é necessário ajustar a dose oral em pacientes com insuficiência renal de grau leve a grave.
O voriconazol é hemodialisável com um clearance de 121 mL/min. Uma sessão de hemodiálise com a duração de 4 horas não remove uma quantidade de voriconazol suficiente que justifique um ajuste posológico.
Uso em Pacientes com Insuficiência Hepática
Não é necessário ajuste de dose em pacientes com comprometimento hepático agudo, manifestado por elevação da função hepática detectada por testes (TGP/ALT, TGO/AST). Recomenda-se a monitoração contínua dos testes da função hepática para verificar elevações posteriores.
Para pacientes com cirrose hepática de grau leve a moderado (classe A e B de Child-Pugh), em tratamento com Voriconazol, recomenda-se o uso dos regimes de dose de ataque padrão, mas somente metade da dose de manutenção.
Voriconazol não foi estudado em pacientes com cirrose hepática crônica grave (classe C de Child-Pugh).
Voriconazol foi associado a elevações dos testes da função hepática e a sinais clínicos de lesão hepática, tal como icterícia, e deve apenas ser utilizado em pacientes com insuficiência hepática grave somente quando o benefício superar o risco potencial. Os pacientes com insuficiência hepática grave devem ser cuidadosamente monitorados quanto à toxicidade do fármaco.
Uso em Crianças
Uso em Crianças (2 a < 12 anos) e Adolescentes (12 a 14 anos e < 50 kg)
O regime de dose recomendado é o seguinte:
| - | Oral |
|
Dose de ataque (nas primeiras 24 horas) |
Não recomendado |
|
Dose de manutenção (após as primeiras 24 horas) |
9 mg/kg a cada 12 horas (dose máxima de 350 mg a cada 12 horas) |
Nota: Baseado na análise de farmacocinética populacional em 112 pacientes pediátricos imunocomprometidos de 2 a <12 anos e 26 adolescentes imunocomprometidos de 12 a <17 anos.
Recomenda-se iniciar a terapia com o regime intravenoso e, o regime oral deve ser considerado somente após uma melhora clínica significante. Foi observado que uma dose intravenosa de 8 mg/kg fornecerá uma exposição ao Voriconazol aproximadamente 2 vezes maior que a dose oral de 9 mg/kg.
A dose oral recomendada para crianças é baseada em estudos onde Voriconazol foi administrado na forma de pó para suspensão oral. Não foi investigado em pacientes pediátricos a bioequivalência entre o pó para suspensão oral e comprimidos.
Considerando o limite assumido gastroentérico de tempo de trânsito em pacientes pediátricos, a absorção dos comprimidos pode ser diferente em pacientes pediátricos e adultos.
A segurança e a eficácia em pacientes pediátricos menores de 2 anos não foi estabelecida. Portanto, Voriconazol não é recomendado para crianças com menos de 2 anos de idade. O uso em pacientes pediátricos de 2 a <12 anos com insuficiência hepática ou renal não foi estudado.
Uso em todos os outros Adolescentes 12 a 14 anos e ≥ 50 kg; 15 a 16 anos independente do peso corpóreo)
O regime posológico de Voriconazol deve ser o mesmo indicado para os adultos.
Ajuste de Dose
Em pacientes com resposta inadequada, a dose pode ser aumentada em intervalos de 1 mg/kg (ou intervalos de 50 mg se a dose oral máxima de 350 mg foi usada inicialmente). Se os pacientes não conseguirem tolerar o tratamento, reduzir a dose em intervalos de 1 mg/kg (ou em intervalos de 50 mg se a dose oral máxima de 350 mg foi usada inicialmente).
Este medicamento não deve ser partido, aberto ou mastigado.
Injetável
Voriconazol pó liofilizado para solução injetável deve ser reconstituído e diluído antes da administração por infusão intravenosa. Não administrar por injeção em “bolus”.
Recomenda-se que Voriconazol, pó liofilizado para solução injetável, seja administrado a uma taxa de no máximo 3 mg/kg por hora, durante 1 a 3 horas.
Derivados Sanguíneos e Suplementação eletrolítica
Voriconazol não deve ser infundido concomitantemente com qualquer derivado sanguíneo ou qualquer infusão rápida de suplementação eletrolítica, ainda que as duas infusões estejam correndo em linhas intravenosas separadas (ou cânulas). Os distúrbios eletrolíticos tais como hipocalemia, hipomagnesemia e hipocalcemia devem ser monitorados e corrigidos, se necessário, antes do início e durante o tratamento com Voriconazol.
Solução eletrolítica intravenosa (não concentrada)
Voriconazol pode ser infundido simultaneamente com outras soluções eletrolíticas intravenosas (não concentradas), porém deve ser infundido através de linha separada.
Nutrição Parenteral Total (NPT)
Voriconazol pode ser infundido simultaneamente com nutrição parenteral total, porém deve ser infundido através de linha separada. Se a NTP for infundida através de cateter de múltiplo lúmen, é necessário que a NPT seja administrada utilizando-se um canal diferente daquele utilizado para Voriconazol.
Uso em Adultos
A terapia com Voriconazol, por via intravenosa deve ser iniciada com o regime de dose de ataque especificado, para se obter no Dia 1, concentrações plasmáticas adequadas. O tratamento intravenoso deve continuar por pelo menos 7 dias antes da troca para a terapia oral. Uma vez que o paciente está clinicamente melhor e torna-se tolerante a medicação administrada por via oral, o comprimido de voriconazol pode ser utilizado. Devido à alta biodisponibilidade oral (96%), a troca entre a administração intravenosa e a oral é adequada, quando indicada clinicamente.
Informações detalhadas das recomendações de dosagem são apresentadas na tabela a seguir:
|
Infecção |
Dose de Ataque (nas primeiras 24 horas) |
Dose de Manutençãoa |
|
Aspergilose invasivab |
6 mg/kg a cada 12 horas |
4 mg/kg a cada 12 horas |
|
Infecções invasivas graves por Candida, inclusive candidemia |
6 mg/kg a cada 12 horas |
3-4 mg/kg a cada 12 horasc |
|
Candidíase esofágica |
6 mg/kg a cada 12 horas |
Não recomendado (utilizar tratamento oral se possível) |
|
Scedosporioses e Fusarioses |
6 mg/kg a cada 12 horas |
4 mg/kg a cada 12 horas |
a. Em estudos com voluntários sadios, a dose oral de 200 mg a cada 12 horas resultou em uma exposição (AUCτ) similar à dose IV de 3 mg/kg a cada 12 horas, a dose oral de 300 mg a cada 12 horas resultou em uma exposição (AUCτ) similar à dose IV de 4 mg/kg a cada 12 horas.
b. Em um estudo clínico pivotal de aspergilose invasiva, a duração mediana do tratamento de voriconazol intravenoso foi de 10 dias (variação de 2 – 85 dias). A duração mediana da terapia oral de voriconazol foi de 76 dias (variação 2 – 232 dias).
c. Em estudos clínicos, pacientes com candidemia receberam como terapia inicial 3 mg/kg a cada 12 horas, enquanto pacientes com outras infecções profundas por Candida receberam 4 mg/kg como terapia de salvamento. Dose apropriada deve basear-se na gravidade e natureza da infecção.
Ajuste de Dose
Se os pacientes não tolerarem o tratamento de 4 mg/kg a cada 12 horas, reduzir a dose intravenosa de manutenção para o mínimo de 3 mg/kg a cada 12 horas.
A fenitoína pode ser coadministrada com voriconazol, se a dose de manutenção do Voriconazol for aumentada para 5 mg/kg por via intravenosa, a cada 12 horas.
Quando Voriconazol é coadministrado com doses ajustadas de efavirenz, a dose de manutenção de Voriconazol deve ser aumentada para 400 mg a cada 12 horas.
A duração do tratamento depende da resposta clínica e micológica dos pacientes. A duração do tratamento com a formulação intravenosa não deve ser superior a 6 meses.
Uso em Pacientes Idosos
Não é necessário ajuste da dose em pacientes idosos.
Uso em Pacientes com Insuficiência Renal
Em pacientes com insuficiência renal moderada a grave (clearance de creatinina < 50 mL/min) ocorre acúmulo do veículo utilizado na formulação intravenosa, SBECD (sulfobutil-éter β-ciclodextrina sódica). Nestes pacientes deve ser administrada a formulação oral de Voriconazol, exceto quando a avaliação de risco-benefício para o paciente justifique o uso da formulação intravenosa. As concentrações séricas de creatinina devem ser rigorosamente monitoradas nestes pacientes e se forem verificados aumentos, deve ser considerada a mudança para tratamento por via oral.
O voriconazol é hemodialisável com um clearance de 121 mL/min. Uma sessão de hemodiálise com a duração de 4 horas não remove uma quantidade de voriconazol suficiente que justifique um ajuste posológico.
O veículo intravenoso, SBECD, é hemodialisável com um clearance de 55 mL/min.
Uso em Pacientes com Insuficiência Hepática
Não é necessário ajuste de dose em pacientes com comprometimento hepático agudo, manifestado por elevação da função hepática detectada por testes (TGP/ALT, TGO/AST). Recomenda-se a monitoração contínua dos testes da função hepática para verificar elevações posteriores.
Para pacientes com cirrose hepática de grau leve a moderado (classe A e B de Child-Pugh), em tratamento com Voriconazol, recomenda-se o uso dos regimes de dose de ataque padrão, mas somente metade da dose de manutenção.
Voriconazol não foi estudado em pacientes com cirrose hepática crônica grave (classe C de Child-Pugh).
Voriconazol foi associado a elevações dos testes da função hepática e a sinais clínicos de lesão hepática, tal como icterícia e deve apenas ser utilizado em pacientes com insuficiência hepática grave somente quando o benefício superar o risco potencial. Os pacientes com insuficiência hepática grave devem ser cuidadosamente monitorados quanto à toxicidade do fármaco.
Uso em Crianças
Uso em Crianças (2 a < 12 anos) e Adolescentes (12 a 14 anos e < 50 kg)
O regime de dose recomendado é o seguinte:
| - |
Intravenosa |
|
Dose de ataque (nas primeiras 24 horas) |
9 mg/kg a cada 12 horas |
|
Dose de manutenção (após as primeiras 24 horas) |
8 mg/kg a cada 12 horas |
Nota: Baseado na análise de farmacocinética populacional em 112 pacientes pediátricos imunocomprometidos de 2 a <12 anos e 26 adolescentes imunocomprometidos de 12 a <17 anos.
Recomenda-se iniciar a terapia com o regime intravenoso e, o regime oral deve ser considerado somente após uma melhora clínica significante. Foi observado que uma dose intravenosa de 8 mg/kg fornecerá uma exposição ao Voriconazol aproximadamente 2 vezes maior que a dose oral de 9 mg/kg.
A segurança e a eficácia em pacientes pediátricos menores de 2 anos não foi estabelecida (vide item 3. Características Farmacológicas – Propriedades Farmacodinâmicas). Portanto, Voriconazol não é recomendado para crianças com menos de 2 anos de idade. O uso em pacientes pediátricos de 2 a <12 anos com insuficiência hepática ou renal não foi estudado.
Uso em todos os outros Adolescentes (12 a 14 anos e ≥ 50 kg; 15 a 16 anos independente do peso corpóreo)
O regime posológico de Voriconazol deve ser o mesmo indicado para os adultos.
Ajuste de Dose
Em pacientes com resposta inadequada, a dose pode ser aumentada em intervalos de 1 mg/kg (ou intervalos de 50 mg se a dose oral máxima de 350 mg foi usada inicialmente). Se os pacientes não conseguirem tolerar o tratamento, reduzir a dose em intervalos de 1 mg/kg (ou em intervalos de 50 mg se a dose oral máxima de 350 mg foi usada inicialmente).
Instruções para Administração
Voriconazol, pó liofilizado para solução injetável, é apresentado em frasco-ampola para uso único e qualquer solução não utilizada deve ser descartada. O conteúdo do frasco deve ser reconstituído com 19 mL de água para injetáveis, obtendo uma solução cristalina contendo 10 mg/mL de voriconazol e um volume extraível de 20 mL. Descarte o frasco-ampola de Voriconazol caso o vácuo não empurre o diluente para dentro do frasco. Antes da administração, o volume de solução reconstituída (vide tabela adiante) deve ser adicionado a um diluente de infusão compatível, recomendado a seguir, para produzir, quando apropriado, uma solução final de Voriconazol equivalente a 0,5-5 mg/mL de voriconazol.
Volumes Requeridos da solução reconstituída de Voriconazol 10 mg/mL
|
Peso Corporal (kg) |
Volume da Solução Reconstituída de Voriconazol (10 mg/mL) necessária para: |
||||
|
Dose de 3 mg/kg (número de frascos-ampola) |
Dose de 4 mg/kg (número de frascos-ampola) | Dose de 6 mg/kg (número de frascos-ampola) | Dose de 8 mg/kg (número de frascos-ampola) |
Dose de 9 mg/kg (número de frascos-ampola) |
|
|
10 |
- | 4,0 mL (1) | - | 8,0 mL (1) |
9,0 mL (1) |
| 15 | - | 6,0 mL (1) | - | 12,0 mL (1) |
13,5 mL (1) |
| 20 | - | 8,0 mL (1) | - | 16,0 mL (1) |
18,0 mL (1) |
| 25 | - | 10,0 mL (1) | - | 20,0 mL (1) |
22,5 mL (2) |
| 30 | 9,0 mL (1) | 12 mL (1) | 18 mL (1) | 24,0 mL (2) |
27,0 mL (2) |
| 35 | 10,5 mL (1) | 14 mL (1) | 21 mL (2) | 28,0 mL (2) |
31,5 mL (2) |
| 40 | 12,0 mL (1) | 16 mL (1) | 24 mL (2) | 32,0 mL (2) |
36,0 mL (2) |
| 45 | 13,5 mL (1) | 18 mL (1) | 27 mL (2) | 36,0 mL (2) |
40,5 mL (3) |
| 50 | 15,0 mL (1) | 20 mL (1) | 30 mL (2) | 40,0 mL (2) |
45,0 mL (3) |
| 55 | 16,5 mL (1) | 22 mL (2) | 33 mL (2) | 44,0 mL (3) |
49,5 mL (3) |
| 60 | 18,0 mL (1) | 24 mL (2) | 36 mL (2) | 48,0 mL (3) |
54,0 mL (3) |
| 65 | 19,5 mL (1) | 26 mL (2) |
39 mL (2) |
52,0 mL (3) |
58,5 mL (3) |
| 70 | 21,0 mL (2) | 28 mL (2) |
42 mL (3) |
- | - |
| 75 | 22,5 mL (2) | 30 mL (2) |
45 mL (3) |
- | - |
| 80 | 24,0 mL (2) | 32 mL (2) |
48 mL (3 |
- | - |
| 85 | 25,5 mL (2) | 34 mL (2) |
51 mL (3) |
- | - |
| 90 | 27,0 mL (2) | 36 mL (2) |
54 mL (3) |
- | - |
| 95 | 28,5 mL (2) | 38 mL (2) |
57 mL (3) |
- | - |
| 100 | 30,0 mL (2) | 40 mL (2) |
60 mL (3) |
- | - |
Voriconazol, pó liofilizado para solução injetável, após reconstituição e diluição, destina-se à administração por infusão intravenosa. Voriconazol não deve ser administrado como injeção em “bolus” ou injeção intramuscular.
Recomenda-se que Voriconazol, pó liofilizado para solução injetável, seja administrado a uma velocidade de infusão máxima equivalente a 3 mg/kg por hora, durante 1 a 2 horas.
Reconstituição
Preparar a solução inicial de Voriconazol, pó liofilizado para solução injetável, adicionando 19 mL de água para injetáveis ao frasco com o pó com 200 mg e agitar até completa dissolução. Cada mL da solução reconstituída contém 10 mg de voriconazol.
Os medicamentos para administração parenteral devem ser inspecionados visualmente quanto à presença de micropartículas antes da administração. Se houver evidência de micropartículas nos líquidos reconstituídos, a solução deve ser descartada.
Diluir essa solução imediatamente antes da administração.
Diluição
A solução reconstituída é compatível e pode ser diluída com as seguintes soluções:
- Cloreto de sódio 9 mg/mL (0,9%) para Infusão Intravenosa;
- Ringer lactato para infusão intravenosa;
- Glicose 5% e ringer lactato de sódio para infusão intravenosa;
- Glicose 5% e cloreto de sódio a 0,45% para Infusão Intravenosa;
- Glicose 5% para Infusão Intravenosa;
- Glicose 5% em 20 mEq de cloreto de potássio para Infusão Intravenosa;
- Cloreto de sódio 0,45% para Infusão Intravenosa;
- Glicose 5% e cloreto de sódio a 0,9% para Infusão Intravenosa.
A compatibilidade de voriconazol com outros diluentes que não os descritos acima, é desconhecida.
Incompatibilidades
Derivados sanguíneos e Suplementação eletrolítica
Voriconazol não deve ser infundido simultaneamente com qualquer derivado sanguíneo ou qualquer infusão rápida de suplementação eletrolítica, ainda que as duas infusões estejam correndo em linhas separadas (ou cânulas). Distúrbios eletrolíticos tais como hipocalemia, hipomagnesemia e hipocalcemia devem ser corrigidas antes do início do tratamento com Voriconazol.
Solução eletrolítica intravenosa (não concentrada)
Voriconazol pode ser infundido simultaneamente com outras soluções eletrolíticas intravenosas (não concentradas), porém deve ser infundido através de linha separada.
Nutrição Parenteral Total (NPT)
Voriconazol pode ser infundido simultaneamente com nutrição parenteral total, porém deve ser infundido através de linha separada. Se a NTP for infundida através de cateter de múltiplo lúmen, é necessário que a NTP seja administrada utilizando-se um canal diferente daquele utilizado para o voriconazol.
Voriconazol não deve ser diluído com infusão intravenosa de bicarbonato de sódio a 4,2%. A compatibilidade com outras concentrações é desconhecida.
Este medicamento não deve ser misturado com outros medicamentos, exceto aqueles mencionados no item “Diluição”.
Não adicionar medicação suplementar (exceto aquelas citadas no item “diluição”) ou utilizar a mesma linha intravenosa para administração de outra medicação simultaneamente.
Quais cuidados devo ter ao usar o Voriconazol Farma Vision Importadora e Exportadora de Medicamentos?
Comprimido / Injetável
Hipersensibilidade
A prescrição de Voriconazol a pacientes com hipersensibilidade a outros agentes azólicos deve ser feita com cautela.
Cardiovascular
Alguns agentes azólicos, incluindo voriconazol, estão associados ao prolongamento do intervalo QT no eletrocardiograma. Foram relatados casos raros durante o desenvolvimento clínico e estudos pós-comercialização de torsade de pointes em pacientes em tratamento com Voriconazol que apresentavam fatores de risco, tais como histórico de quimioterapia cardiotóxica, cardiomiopatia, hipocalemia e em tratamento com medicações concomitantes que podem contribuir.
Voriconazol deve ser administrado com cautela a pacientes com condições potenciais para o desenvolvimento de pró-arritmias tais como:
- Prolongamento QT congênito ou adquirido;
- Cardiomiopatia, em particular quando há insuficiência cardíaca;
- Bradicardia sinusal;
- Arritmias sintomáticas existentes;
- Medicamentos concomitantes conhecidos por prolongar o intervalo QT.
Distúrbios eletrolíticos, como hipocalemia, hipomagnesemia e hipocalcemia, devem ser monitorados e corrigidos, se necessário, antes do início e durante a terapia com Voriconazol.
Foi conduzido um estudo em voluntários sadios que investigou o efeito no intervalo QT de doses únicas de voriconazol até 4 vezes a dose diária usual. Nenhum indivíduo dos grupos apresentou um aumento no intervalo QTc ≥ 60 ms em relação ao pré-tratamento. Nenhum voluntário apresentou um intervalo que excedeu o limiar de potencial de relevância clínica de 500 ms.
Toxicidade hepática
Nos estudos clínicos houve casos de reações hepáticas graves durante o tratamento com Voriconazol (envolvendo hepatite clínica, colestase e insuficiência hepática fulminante, incluindo morte). Foram observados casos de reações hepáticas principalmente em pacientes com condições clínicas subjacentes graves (predominantemente com doença hematológica maligna). Ocorreram reações hepáticas transitórias, incluindo hepatite e icterícia em pacientes sem outros fatores de risco identificáveis. A disfunção hepática foi geralmente reversível com a descontinuação do tratamento.
Monitoramento da função hepática
Pacientes que estejam recebendo Voriconazol devem ser cuidadosamente monitorados quanto à toxicidade hepática. O acompanhamento clínico deve incluir avaliação laboratorial da função hepática (especialmente AST e ALT) no início do tratamento com Voriconazol e pelo menos semanalmente no primeiro mês do tratamento. Se o tratamento for continuado, a frequência do monitoramento poderá ser reduzida para uma vez por mês se não houver alterações nos testes da função hepática.
Se os testes da função hepática passarem a apresentar valor notavelmente alto, Voriconazol deverá ser descontinuado, a não ser que a avaliação médica dos riscos e benefícios do tratamento para o paciente justifique seu uso continuado.
Eventos adversos visuais
Há relatos na pós-comercialização de eventos adversos visuais prolongados, incluindo neurites ópticas e papiledema. Estes eventos ocorreram principalmente em pacientes com doenças graves que possuíam comorbidades e/ou medicações concomitantes que causaram ou contribuíram com estes eventos.
Eventos adversos renais
Foi observada insuficiência renal aguda em pacientes em estado grave submetidos ao tratamento com Voriconazol. Pacientes sendo tratados com voriconazol podem também ser tratados com medicamentos nefrotóxicos e ter condições concomitantes que podem resultar em diminuição da função renal.
Monitoramento da função renal
Os pacientes devem ser monitorados quanto ao desenvolvimento de alterações na função renal. A monitoração deve incluir avaliação laboratorial, particularmente da creatinina sérica.
Monitoramento da função pancreática
Adultos e crianças com fatores de risco para pancreatite aguda (p. ex. quimioterapia recente, transplante de células tronco hematopoiéticas) devem ser monitorados quanto ao desenvolvimento de pancreatite durante tratamento com voriconazol.
Eventos adversos dermatológicos
Os pacientes desenvolvem reações cutâneas esfoliativas, tais como síndrome de Stevens-Johnson, durante o tratamento com Voriconazol. Se o paciente desenvolver reação cutânea esfoliativa, Voriconazol deve ser descontinuado.
Além disso, Voriconazol foi associado a reações de fotossensibilidade cutânea. Recomenda-se que os pacientes, incluindo crianças, evitem a exposição à luz solar direta durante o tratamento com Voriconazol e usem medidas como roupas de proteção e filtro solar com alto fator de proteção solar (FPS).
Tratamento de longo prazo
Os seguintes eventos adversos graves referentes ao tratamento de longo prazo com Voriconazol foram relatados.
Carcinoma de pele de células escamosas (CCE)
Em pacientes com reações cutâneas devido à fotossensibilidade e fatores de risco adicionais, carcinoma de pele de células escamosas e melanoma foram relatados durante terapias de longo prazo. Caso ocorram reações fototóxicas, deve-se buscar aconselhamento multidisciplinar e o paciente deve ser encaminhado a um dermatologista. A descontinuação de Voriconazol deve ser considerada. Avaliações dermatológicas devem ser realizadas de forma sistemática e regular sempre que o voriconazol for continuado apesar da ocorrência de lesões relacionadas à fototoxicidade, de forma a permitir a detecção antecipada e o gerenciamento de lesões pré-malignas. Se o paciente desenvolver lesão cutânea compatível com lesões de pele pré-malignas, carcinoma de células escamosas da pele ou melanoma, a descontinuação de Voriconazol deve ser considerada.
Periostite não-infecciosa
Periostite foi reportada em pacientes transplantados durante o tratamento de longo prazo com voriconazol. Se um paciente desenvolver dor esquelética e achados radiológicos compatíveis com periostite, voriconazol deve ser descontinuado.
Uso pediátrico
A segurança e a eficácia em pacientes pediátricos com idade inferior a 2 anos ainda não foram estabelecidas. Voriconazol é indicado para pacientes pediátricos com idade superior a 2 anos de idade. A maior frequência de elevações de enzimas hepáticas foi observada na população pediátrica (vide item 9. Reações adversas). A função hepática deve ser monitorada tanto em crianças quanto em adultos. A bioequivalência oral pode ser limitada em pacientes pediátricos de 2 a 12 anos com má-absorção e com peso muito baixo para a idade. Nestes casos, a administração intravenosa de Voriconazol é recomendada.
A frequência das reações de fototoxicidade é mais alta na população pediátrica. Uma vez que uma evolução para CCE foi relatada, medidas rigorosas de fotoproteção são justificadas para essa população de pacientes. Em crianças com lesões de fotoenvelhecimento, como lentigo ou nevus, recomenda-se evitar exposição ao sol e acompanhamento dermatológico mesmo após a descontinuação do tratamento.
Everolimo (substrato da CYP3A4, substrato da P-gp)
A coadministração de voriconazol com everolimo não é recomendada, pois voriconazol pode aumentar significativamente as concentrações de everolimo. Atualmente existem dados insuficientes para permitir recomendações posológicas nesta situação.
Fluconazol (inibidor da CYP2C9, CYP2C19 e CYP3A4)
A coadministração de voriconazol oral e fluconazol oral resultou em um aumento significativo na Cmáx e na AUCT de voriconazol em sujeitos sadios. A redução da dose e/ou da frequência de voriconazol e de fluconazol que poderia eliminar este efeito não foi estabelecida. Recomenda-se a monitoração de eventos adversos associados com voriconazol se voriconazol for utilizado em seguida a fluconazol.
Efavirenz (indutora do CYP450, inibidor e substrato do CYP3A4)
Quando Voriconazol é coadministrado com efavirenz a dose de voriconazol deve ser aumentada para 400 mg a cada 12 horas e a dose de efavirenz deve ser diminuída para 300 mg a cada 24 horas.
Fenitoína (substrato do CYP2C9 e potente indutora do CYP450)
Recomenda-se a monitoração cuidadosa das concentrações de fenitoína, quando esta for coadministrada com voriconazol. O uso concomitante de ambos deve ser evitado, a menos que o benefício supere o risco.
Ritonavir (potente indutor CYP450, inibidor e substrato da CYP3A4)
Coadministração de voriconazol e baixas doses de ritonavir (100 mg a cada 12 horas) deve ser evitada a menos que uma avaliação do risco/benefício justifique o uso de Voriconazol.
Metadona (substrato da CYP3A4)
O aumento da concentração plasmática da metadona foi associado com toxicidade incluindo prolongamento do intervalo QT. É recomendado durante a coadministração o frequente monitoramento dos eventos adversos e da toxicidade da metadona. A redução da dose da metadona pode ser necessária.
Opioides de ação curta (substrato de CYP3A4)
A redução na dose da alfentanila, fentanila e outros opioides de ação curta com estrutura similar a alfentanila e metabolizados pelo CYP3A4 (p. ex. sufentanila) deve ser considerada quando coadministrado com voriconazol. Como a meiavida da alfentanila é prolongada em quatro vezes quando a alfentanila é coadministrada com voriconazol, e em um estudo publicado independente, o uso concomitante de voriconazol com fentanila resultou em um aumento de 1,4 vezes da AUC0-∞ média de fentanila, pode ser necessária uma frequente monitoração das reações adversas associadas aos opioides (incluindo período prolongado de monitoração respiratória).
Opioides de ação longa (substrato do CYP3A4)
A redução na dose de oxicodona e outros opioides de ação longa metabolizados pelo CYP3A4 (p. ex. hidrocodona) deve ser considerada quando coadministrado com Voriconazol. Pode ser necessária uma frequente monitoração das reações adversas associadas aos opioides.
Fertilidade, Gravidez e Lactação
Gravidez
Não estão disponíveis informações adequadas sobre a utilização de Voriconazol em mulheres grávidas.
Os estudos em animais mostraram toxicidade reprodutiva em altas doses. O risco potencial para seres humanos é desconhecido.
Voriconazol não deve ser utilizado durante a gravidez, a menos que o benefício para a mãe supere claramente o risco potencial para o feto.
Voriconazol é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Mulheres com Potencial para Engravidar
As mulheres com potencial para engravidar devem sempre utilizar um método contraceptivo eficaz durante o tratamento.
Lactação
A excreção do voriconazol no leite materno não foi investigada. A amamentação deve ser interrompida ao iniciar o tratamento com Voriconazol.
Fertilidade
Em um estudo em animais não foi demonstrado comprometimento da fertilidade em ratos machos e fêmeas.
Efeitos na Habilidade de Dirigir e Operar Máquinas
O voriconazol pode causar alterações transitórias e reversíveis na visão, incluindo visão embaçada, aumento ou alteração da percepção visual e/ou fotofobia. Os pacientes devem evitar as tarefas potencialmente perigosas, tais como dirigir ou operar máquinas, enquanto estiverem apresentando estes sintomas. Os pacientes não devem dirigir à noite durante o tratamento com Voriconazol.
Exclusivo Comprimido
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
Exclusivo Injetável
Reações relacionadas com a infusão
Durante a administração da formulação intravenosa de voriconazol foram observadas reações relacionadas com a infusão, predominantemente rubor e náuseas. Dependendo da gravidade dos sintomas deve-se considerar a interrupção do tratamento
Quais as reações adversas e os efeitos colaterais do Voriconazol Farma Vision Importadora e Exportadora de Medicamentos?
Comprimido / Injetável
O perfil de segurança do voriconazol em adultos está baseado em um banco de dados de segurança integrado composto de mais de 2000 indivíduos (1.603 pacientes adultos em estudos terapêuticos). Isto representa uma população heterogênea, abrangendo pacientes com doença hematológica maligna, pacientes infectados por HIV com candidíase esofágica e infecções fúngicas refratárias, pacientes não neutropênicos com candidemia ou aspergilose e voluntários sadios.
Os eventos adversos mais comumente relatados foram os distúrbios visuais, teste de função hepática anormal, febre, rash, vômitos, náusea, diarreia, cefaleia, edema periférico e dor abdominal. A gravidade dos eventos adversos foi geralmente de leve a moderada. Não foram observadas diferenças clinicamente significativas na análise dos dados de segurança por idade, raça ou sexo.
As reações adversas pela classe de sistema de órgãos e a frequência de categoria CIOMS (Council for International Organizations of Medical Sciences) estão listadas em ordem decrescente de gravidade médica dentro de cada frequência de categoria e classe de sistema de órgãos:
|
Classe de Sistema de Órgãos |
Muito Comum ≥ 1/10 | Comum ≥ 1/100 a < 1/10 | Incomum ≥ 1/1.000 a < 1/100 | Raro ≥ 1/10.000 a < 1/1.000 |
Frequência não conhecida (não pode ser estimada a partir dos dados disponíveis) |
|
Infecções e infestações |
- | Sinusite | Colite pseudomenbranosa | - | - |
|
Neoplasias benignas, malignas e não especificadas (incluindo cistos e pólipos) |
- | - | - | - |
Ccarcinoma de células escamosas*,g |
|
Distúrbios do sistema linfático e sangue |
- | Agranulocitosea , pancitopenia, trombocitopeniab , leucopenia, anemia | Insuficiência de medula óssea, linfadenopatia, eosinofilia | Coagulação intravascular disseminada | - |
|
Distúrbios do sistema imune |
- | - | Hipersensibilidade | Reação anafilactoide | - |
| - | - | Insuficiência adrenal, hipotireoidismo | Hipertireoidismo | - | |
|
Distúrbios do metabolismo e nutrição |
Edema periférico | Hipoglicemia, hipocalemia, hiponatremia* | - | - | - |
|
Distúrbios psiquiátricos |
- | Depressão, alucinação, ansiedade, insônia, agitação, estado de confusão | - | - | - |
|
Distúrbios do sistema nervoso |
Dor de cabeça | Síncope, tremor, hipertoniae , parestesia, sonolência, tontura | Edema cerebral, encefalopatiac , distúrbios extrapiramidaisd , neuropatia periférica, ataxia, hipoestesia, disgeusia | Encefalopatia hepática, síndrome de Guillain-Barré, nistagmo | - |
|
Distúrbios visuais |
Distúrbio visualh | Hemorragia da retina | Papiledemag , crise oculogírica, diplopia, esclerite, blefarite | Atrofia óptica, distúrbio do nervo ópticof , opacidade da córnea | - |
|
Distúrbios do ouvido e labirinto |
- | - | Hipoacusia, vertigem, zumbido | - | - |
|
Distúrbios cardíacos |
- | Arritmia supraventricular, taquicardia, bradicardia | Fibrilação ventricular, extrasístole ventricular, taquicardia ventricular, prolongamento QT no eletrocardiograma, taquicardia supraventricular | Torsades de pointes, bloqueio atrioventricular completo, bloqueio de ramo, ritmo nodal | - |
|
Distúrbios vasculares |
- | Hipotensão, flebite | Tromboflebite, linfangite | - | - |
|
Distúrbios respiratório, torácico e mediastinal |
- | Síndrome do desconforto respiratório agudo, edema pulmonar | - | - | - |
|
Distúrbios gastrointestinais |
Diarreia, vômito, náusea | Queilite, dispepsia, dor abdominal, constipação, gengivite | Peritonite, pancreatite, língua inchada, duodenite, gastroenterite, glossite | - | - |
| Teste de função hepática anormal | Icterícia, icterícia colestática | Insuficiência hepática, hepatitei , hepatomegalia, colecistite, colelitíase | - | - | |
|
Distúrbios da pele e tecidos subcutâneos |
Rash | Dermatite esfoliativa, alopecia, púrpura, rash, máculo-papular, prurido | Síndrome de Stevens-Johnson, reação de fotossensibilidade, urticária | Necrólise epidérmica tóxica, angioedema, pseudoporfiria, eritema multiforme, psoríase, erupção medicamentosa, eczema |
Lúpus eritematoso cutâneo* |
|
Distúrbios musculoesquelé tico e do tecido conjuntivo |
- | Dor nas costas | Artrite | - | - |
|
Distúrbios renal e urinário |
- | Insuficiência renal aguda, hematúria | Necrose tubular renal, proteinúria, nefrite | - | - |
|
Distúrbios gerais e alterações no local de administração |
Pirexia | Dor no peito, edemaj da face, astenia, calafrios | Sintomas de gripe | - | - |
|
Testes laboratoriais |
- | Creatinina sanguínea elevada | Ureia no sangue aumentada, colesterol sanguíneo elevado | - | - |
*Reações adversas identificadas pós comercialização.
a Inclui neutropenia febril e neutropenia.
b Inclui púrpura trombocitopênica imune.
c Inclui encefalopatia hipóxico-isquêmica e encefalopatia metabólica. d Inclui acatisia e parkinsonismo. e Inclui rigidez de nuca e tétano.
f Neurite óptica prolongada foi notificado na pós-comercialização. Vide item 5. Advertências e Precauções.
g Vide item Quais cuidados devo ter ao usar o Voriconazol?.
h Vide "distúrbios visuais" no item Quais as reações adversas e os efeitos colaterais do Voriconazol?.
i Inclui lesão hepática induzida por medicamentos, hepatite tóxica, lesão hepatocelular e hepatotoxicidade.
j Inclui edema periorbital, edema de lábios e edema de boca.
Distúrbios Visuais
Em estudos clínicos, distúrbios visuais (incluindo visão turva, fotofobia, cloropsia, cromatopsia, daltonismo, cianopsia, distúrbio ocular, visão de halo, cegueira noturna, oscilopsia, fotopsia, escotoma cintilante, acuidade visual reduzida, brilho visual, defeito no campo visual, flocos vítreos, e xantopsia) com voriconazol foram muito comuns.
Estes distúrbios visuais foram temporários e totalmente reversíveis, sendo que a maioria foi resolvida espontaneamente dentro de 60 minutos. Houve evidência de atenuação com doses repetidas de voriconazol. Os distúrbios visuais foram geralmente leves, resultando raramente em descontinuação do tratamento e não foram associados a sequelas em longo prazo. Os distúrbios visuais podem estar associados aos níveis plasmáticos e/ou doses mais elevadas.
Há relatos de eventos visuais prolongados no período pós-comercialização.
O mecanismo de ação é desconhecido, embora o local de ação mais provável seja dentro da retina.
Num estudo realizado em voluntários sadios em que foi analisado o impacto do voriconazol sobre a função da retina, verificou-se que o voriconazol causou diminuição da amplitude das ondas do eletroretinograma (ERG). O ERG permite medir as correntes elétricas na retina. As alterações do ERG não progrediram ao longo dos 29 dias de tratamento e foram totalmente revertidas com a descontinuação do tratamento com voriconazol.
O efeito em longo prazo de voriconazol (média de 169 dias; variando de 5-353 dias) na função visual foi avaliado em indivíduos com paracoccidioidomicose. O voriconazol não apresentou efeitos clinicamente relevantes na função visual conforme avaliado por testes de acuidade visual, campos visuais, cores visuais e sensibilidade de contraste. Não houve sinais de toxicidade na retina. Dezessete dos 35 pacientes tratados com voriconazol apresentaram eventos adversos visuais. Estes eventos não levaram à descontinuação do medicamento; foram geralmente leves, ocorreram durante a primeira semana de tratamento e desapareceram durante o tratamento contínuo com voriconazol.
Reações Dermatológicas
Reações dermatológicas foram muito comuns nos pacientes tratados com voriconazol nos estudos clínicos, porém, estes pacientes apresentavam doenças de base graves e estavam recebendo medicações múltiplas concomitantes. A gravidade da maioria dos rashes (erupções cutâneas) foi de leve à moderada. Pacientes desenvolveram reações cutâneas graves, incluindo síndrome de Stevens-Johnson (incomum), necrólise epidérmica tóxica (raro) e eritema multiforme (raro), durante o tratamento com Voriconazol.
Caso os pacientes desenvolvam rash cutâneo, eles devem ser monitorados cuidadosamente e Voriconazol deve ser descontinuado se as lesões progredirem. Foram relatadas reações cutâneas de fotossensibilidade especialmente em tratamentos de longo prazo.
Reações adversas dermatológicas potencialmente relacionadas com a fototoxicidade (pseudoporfiria, queilite, e lúpus eritematoso cutâneo) também foram relatadas com voriconazol. Evitar exposição ao sol e fotoproteção são recomendados para todos os pacientes. Se ocorrer fototoxicidade, a descontinuação do voriconazol e avaliação dermatológica devem ser consideradas.
Testes de Função Hepática
A incidência geral de aumento das transaminases > 3 x LSN (não necessariamente compreendendo um evento adverso) no programa clínico do voriconazol foi de 18,0% (319/1.768) em indivíduos adultos e 25,8% (73/283) em pacientes pediátricos que receberam voriconazol para uso terapêutico e profilático combinados. As anormalidades nos testes de função hepática podem estar associadas ao aumento das concentrações plasmáticas e/ou doses. A maioria dos testes de função hepática anormal foi resolvida ou durante o tratamento, sem ajuste da dose, ou após um ajuste da dose, incluindo a descontinuação da terapia.
O voriconazol tem sido associado com casos de toxicidade hepática grave em pacientes com outras condições graves de base. Isto inclui casos de icterícia, hepatite e insuficiência hepática que ocasionaram óbito.
Uso Pediátrico
A segurança de voriconazol foi analisada em 285 pacientes pediátricos com idade variando de 2 a < 12 anos, tratados com voriconazol em estudos farmacocinéticos (127 pacientes pediátricos) e em programa de uso por compaixão (158 pacientes pediátricos). O perfil das reações adversas dos 285 pacientes pediátricos foi similar ao dos adultos. Os dados pós-comercialização sugerem que pode haver maior ocorrência de reações de pele na população pediátrica quando comparada aos adultos.
Houve relatos pós-comercialização de pancreatite em pacientes pediátricos.
Em casos de eventos adversos, notifique ao Sistema de Notificação de Eventos Adversos a Medicamentos - Vigimed, disponível em http://portal.anvisa.gov.br/vigimed, ou para a Vigilância Sanitária Estadual ou Municipal.
Exclusivo Injetável
Reações Relacionadas com a Infusão
Durante a infusão da formulação intravenosa do voriconazol em indivíduos sadios, ocorreram reações do tipo anafilactoide, incluindo rubor, febre, transpiração, taquicardia, opressão torácica, dispneia, desmaios, náuseas, prurido e rash cutâneo. Os sintomas surgiram imediatamente após o início da infusão.
Interação medicamentosa: quais os efeitos de tomar Voriconazol Farma Vision Importadora e Exportadora de Medicamentos com outros remédios?
O voriconazol é metabolizado pelas e inibe a atividade das isoenzimas do citocromo P450 CYP2C19, CYP2C9 e CYP3A4. Inibidores ou indutores destas isoenzimas podem aumentar ou diminuir as concentrações plasmáticas de voriconazol, respectivamente, e existe potencial do voriconazol aumentar as concentrações plasmáticas de substâncias metabolizadas por estas isoenzimas do CYP450.
A não ser quando especificado de outro modo, estudos de interações medicamentosas foram realizados em homens adultos sadios usando administrações múltiplas até o estado de equilíbrio com voriconazol oral a 200 mg duas vezes ao dia. Estes resultados são relevantes para outras populações e vias de administração.
O voriconazol deve ser administrado com cuidado em pacientes com medicação concomitante que conhecidamente prolonga o intervalo QT. Quando também houver um potencial de voriconazol aumentar as concentrações plasmáticas de substâncias metabolizadas por isoenzimas da CYP3A4 (certos anti-histamínicos, quinidina, cisaprida, pimozida), a coadministração é contraindicada.
Tabela de interação
Interações entre voriconazol e outros medicamentos são relacionadas na tabela abaixo. (QD = uma vez ao dia, BID = duas vezes ao dia, TID = três vezes ao dia, e ND = não determinado). A direção da seta para cada parâmetro farmacocinético é baseada no intervalo de confiança de 90% da razão da média geométrica, sendo dentro (↔), abaixo (↓) ou acima (↑) da faixa de 80-125%. O asterisco (*) indica uma interação de duas vias. AUCT, AUCT e AUC0-∞ representam a área sob a curva de um intervalo de administração, do tempo zero até o tempo com medição detectável e do tempo zero até infinito, respectivamente.
As interações na tabela são apresentadas na seguinte ordem: contraindicações, aquelas que necessitam de ajuste da dose e monitoração clínica e/ou biológica cuidadosa e finalmente aquelas que não têm interação farmacocinética significativa, mas podem ser de interesse clínico neste campo terapêutico.






Especificações sobre o Voriconazol Farma Vision Importadora e Exportadora de Medicamentos
Caracteristicas Principais
| Fabricante | Farma Vision Importadora e Exportadora de Medicamentos | |
| Tipo do Medicamento | Genérico | |
| Necessita de Receita | Branca Comum (Venda Sob Prescrição Médica) | |
| Princípio Ativo | Voriconazol | |
| Categoria do Medicamento | Antifúngico | |
| Especialidades | Infectologia | |
| Registro no Ministério da Saúde | 1746500120021 | |
| Código de Barras | 619205418260 | |
| Temperatura de Armazenamento | Temperatura ambiente | |
| Produto Refrigerado | Este produto não precisa ser refrigerado | |
| Modo de Uso | Uso injetável (intravenoso) | |
| Pode partir | Esta apresentação não pode ser partida | |
Que tal conferir outros itens? Temos outras opções que podem te interessar!



