Bula do Kaletra
Princípio Ativo:Lopinavir + Ritonavir
Classe Terapêutica:Inibidores Da Protease
Kaletra, para o que é indicado e para o que serve?
Kaletra® (lopinavir/ritonavir) é destinado, em combinação com outros medicamentos antirretrovirais, ao tratamento da infecção pelo Vírus da Imunodeficiência Humana (HIV). A indicação é baseada em análises dos níveis de RNA HIV no plasma (carga viral do HIV no sangue) e células CD4. Até o momento, não há estudos avaliando o efeito do Kaletra® (lopinavir/ritonavir) na progressão da infecção pelo HIV.
Como o Kaletra funciona?
Solução oral / Comprimido revestido
Kaletra® (lopinavir/ritonavir) é um medicamento pertencente à classe dos inibidores de protease, que contém lopinavir e ritonavir e, em combinação com outros agentes antirretrovirais, é indicado para o tratamento de infecção por HIV.
Kaletra® (lopinavir/ritonavir) não cura a infecção por HIV. O medicamento tem por objetivo controlar a quantidade de vírus e promover a melhora do sistema de defesa imunológica do organismo. Kaletra® (lopinavir/ritonavir) reduz a quantidade de HIV no sangue e aumenta o número de células de defesa do organismo.
Durante o tratamento, podem se desenvolver outras infecções, as chamadas oportunistas, ou mesmo outras complicações associadas com a AIDS (Síndrome da Imunodeficiência Adquirida).
O mecanismo de ação do Kaletra® (lopinavir/ritonavir) é inibir a multiplicação do HIV dentro das células, impedindo a ação da enzima protease. A inibição da protease leva à formação de um vírus imaturo, não infeccioso, ou seja, que não é capaz de entrar em outra célula para se multiplicar.
Kaletra® (lopinavir/ritonavir) é um medicamento de uso contínuo e, portanto, assim que atingida a concentração indicada no organismo, o medicamento permanecerá em constante ação.
Exclusivo comprimido revestido
Estudos clínicos demonstraram que a administração de Kaletra® (lopinavir/ritonavir) em pacientes adultos, duas vezes ao dia ou uma única vez ao dia, proporciona eficácia antiviral semelhante. A escolha do intervalo entre as tomadas deve ser orientada pelo médico.
A administração de Kaletra® (lopinavir/ritonavir) na apresentação de 100 mg/25 mg uma única vez ao dia não foi estudada em pacientes pediátricos.
Quais as contraindicações do Kaletra?
Kaletra® (lopinavir/ritonavir) é contraindicado, ou seja, não deve ser usado, em pacientes com hipersensibilidade (alergia) conhecida ao lopinavir/ritonavir ou a qualquer componente presente na formulação.
Kaletra® (lopinavir/ritonavir) não deve ser administrado em combinação a outros medicamentos cujo mecanismo de eliminação seja o mesmo que o seu e, cuja alta concentração no sangue esteja associada a reações adversas graves.
Os medicamentos que não devem ser administrados com Kaletra® (lopinavir/ritonavir) são os seguintes:
- Antagonistas alfa1-adrenoreceptores (cloridrato de alfuzosina), antianginal (ranolazina), antiarrítmicos (dronedarona), antibióticos (ácido fusídico), agentes anticancerígenos (neratinibe, apalutamida), agentes antigotosos (colchicina em pacientes com insuficiência renal e/ou hepática), benzodiazepínicos (midazolam, triazolam), derivados do ergot (ergotamina, di-hidroergotamina, ergonovina e metilergonovina), agentes que atuam na motilidade gastrointestinal (cisaprida), antihistamínicos (astemizol, terfenadina), antipsicóticos (blonanserina, lurasidona e pimozida), produtos herbais (erva-de-São-João – Hypericum perforatum), antivirais de ação direta (DAA) para o tratamento da Hepatite C (elbasvir, grazoprevir), agentes modificadores de lipídios, como inibidores de HMGCoA redutase (lovastatina, sinvastatina) e inibidores da proteína de transferência de triglicerídeos microssomais – MTTP (lomitapida), agonistas de longa duração de beta-adrenoreceptores (salmeterol), inibidores da enzima PDE5 (sildenafila* - somente quando utilizada para tratamento da hipertensão arterial pulmonar).
*Ver seção “Quais cuidados devo ter ao usar o Kaletra?” para uso da sildenafila em pacientes com disfunção erétil.
Como usar o Kaletra?
Solução oral
Kaletra® (lopinavir/ritonavir) solução oral deve ser administrado com alimento.
Utilização de Kaletra® (lopinavir/ritonavir) solução oral com um tubo de alimentação
A dose prescrita de Kaletra® (lopinavir/ritonavir) solução oral pode ser administrada através de um tubo de alimentação. Siga as instruções para o tubo de alimentação para administrar o medicamento.
Produtos que contenham álcool, como Kaletra® (lopinavir/ritonavir), não são recomendados para uso com tubos de alimentação de poliuretano devido ao potencial de incompatibilidade.
Posologia para adultos
- A dose recomendada de Kaletra® (lopinavir/ritonavir) é de 400 mg/100 mg (5,0 mL de solução oral) duas vezes ao dia com alimentação.
Tratamento concomitante - efavirenz, nevirapina, amprenavir ou nelfinavir
- Um aumento da dose de Kaletra® (lopinavir/ritonavir) para 533/133 mg (6,5 mL de solução oral) duas vezes ao dia, administrados com alimentos, deve ser considerado quando usado em combinação com efavirenz, nevirapina, amprenavir ou nelfinavir.
Posologia para crianças (06 meses a 12 anos) - esquema posológico empregando peso do paciente
Para todos os medicamentos, incluindo Kaletra® (lopinavir/ritonavir) solução oral, as quantidades totais de álcool e propilenoglicol que são dadas a crianças, devem ser levadas em consideração, para evitar a toxicidade destes excipientes.
- Em crianças de 06 meses a 12 anos de idade, a dose recomendada de Kaletra® (lopinavir/ritonavir) solução oral é 12/3 mg/kg para as crianças entre 7 kg e 15 kg e de 10/2,5 mg/kg para aquelas com 15 kg a 40 kg, administrada duas vezes ao dia, com alimentos, até a dose máxima de 400 mg/100 mg em crianças acima de 40 kg (5,0 mL de solução oral) duas vezes ao dia.
|
Guia de doses pediátricas baseado no peso sem terapia concomitante |
||
|
Peso |
Dose (mg/kg)* |
Volume de solução oral 2 vezes ao dia (lopinavir 80 mg/mL + ritonavir 20 mg/mL) |
|
7 a < 15 kg |
12/3 mg/kg 2 X dia | - |
|
7 a 10 kg |
- |
1,25 mL |
|
>10 a <15 kg |
- |
1,75 mL |
|
15 a 40 kg |
10/2,5 mg/kg 2 X dia | - |
|
15 a 20 kg |
- |
2,25 mL |
|
> 20 a 25 kg |
- |
2,75 mL |
|
> 25 a 30 kg |
- |
3,50 mL |
|
> 30 a 35 kg |
- |
4,00 mL |
|
> 35 a 40 kg |
- |
4,75 mL |
|
Acima de 40 kg |
Dose do adulto |
5,00 mL |
* Posologia baseada no componente lopinavir da solução lopinavir/ritonavir (80 mg/20 mg por mL).
Observação: utilizar a dose recomendada para adultos em crianças com mais de 12 anos de idade.
Tratamento concomitante - efavirenz, nevirapina, amprenavir ou nelfinavir
- Um aumento da dose de Kaletra® (lopinavir/ritonavir) solução oral para 13/3,25 mg/kg para as crianças com peso entre 7 kg e 15 kg, e para 11/2,75 mg/kg para aquelas com 15 kg a 45 kg, duas vezes ao dia, administrada com alimentos, até a dose máxima de 533/133 mg em crianças com mais de 45 kg, duas vezes ao dia, deve ser considerado quando usado em combinação com efavirenz, nevirapina, amprenavir ou nelfinavir em crianças.
|
Guia de doses pediátricas baseado no peso com terapia concomitante (efavirenz, nevirapina, amprenavir ou nelfinavir) |
||
|
Peso |
Dose (mg/kg)* |
Volume de solução oral 2 vezes ao dia (80 mg lopinavir/20 mg ritonavir por mL) |
|
7 a < 15 kg |
13/3,25 mg/kg 2 X dia | - |
|
7 a 10 kg |
- |
1,50 mL |
|
>10 a <15 kg |
- |
2,00 mL |
|
15 a 45 kg |
11/2,75 mg/kg 2 X dia | - |
|
15 a 20 kg |
- |
2,50 mL |
|
> 20 a 25 kg |
- |
3,25 mL |
|
> 25 a 30 kg |
- |
4,00 mL |
|
> 30 a 35 kg |
- |
4,50 mL |
|
> 35 a 40 kg |
- |
5,00 mL |
|
> 40 a 45 kg |
- |
5,75 mL |
|
Acima de 45 kg |
Dose do adulto |
6,50 mL |
* Posologia baseada no componente lopinavir da solução lopinavir/ritonavir (80 mg/20 mg por mL).
Observação: utilizar a dose recomendada para adultos em crianças com mais de 12 anos de idade.
Posologia para crianças (06 meses a 12 anos) - esquema posológico empregando a área de superfície corporal (m²)
- A dose recomendada de Kaletra® (lopinavir/ritonavir) solução oral para crianças (com 06 meses de idade e acima) é de 230/57,5 mg/m2 duas vezes ao dia, com alimentação, até uma dose máxima de 400 mg/100 mg (5,0 mL) duas vezes ao dia. A dose de 230/57,5 mg/m2 pode ser insuficiente em algumas crianças quando houver administração concomitante a nevirapina, efavirenz, nelfinavir ou amprenavir.
- Um aumento da dose de Kaletra® (lopinavir/ritonavir) solução oral para 300/75 mg/m2 deve ser considerado nesses pacientes. Quando possível, a dose deve ser administrada utilizando uma seringa de dosagem oral calibrada.
A tabela a seguir contém esquemas de doses de Kaletra® (lopinavir/ritonavir) com base na área da superfície corporal.
|
Guia de dose pediátrica baseado na área de superfície corporal sem terapia concomitante |
|
|
Área de superfície corporal (m2)* |
Dose (duas vezes ao dia) |
|
0,25 |
0,7 mL (57,5/14,4 mg) |
|
0,50 |
1,4 mL (115/28,8 mg) |
|
0,75 |
2,2 mL (72,5/43,1 mg) |
|
1,00 |
2,9 mL (230/57,5 mg) |
|
1,25 |
3,6 mL (287,5/71,9 mg) |
|
1,50 |
4,3 mL (345/86,3 mg) |
|
1,75 |
5,0 mL (402,5/100,6 mg) |
* A Área de Superfície Corporal (ASC) pode ser calculada a partir da seguinte equação: Área de Superfície Corporal (m2) = raiz quadrada [altura (cm) X peso (kg) / 3600].
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento.
Não interrompa o tratamento sem o conhecimento do seu médico.
Comprimido
Kaletra® (lopinavir/ritonavir) comprimidos revestidos podem ser tomados com ou sem alimentação.
Posologia para adultos
A dose recomendada de Kaletra® (lopinavir/ritonavir) comprimidos revestidos é:
- Dois comprimidos de 200 mg/50 mg (400 mg/100 mg) duas vezes ao dia com ou sem alimentação, ou
- Quatro comprimidos de 200 mg/50 mg (800 mg/200 mg) uma única vez ao dia com ou sem alimentos, em pacientes sem tratamento prévio ou naqueles com experiência prévia e com menos de três mutações associadas ao lopinavir.
Não há dados suficientes para suportar a administração em dose única diária de Kaletra® (lopinavir/ritonavir) em pacientes com três ou mais mutações associadas ao lopinavir.
Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com carbamazepina, fenobarbital e fenitoína.
A dose única diária é uma alternativa à terapia convencional de dois comprimidos, duas vezes ao dia.
Portanto, o médico decidirá se você deve tomar a dose recomendada por ele uma vez ao dia ou dividí-la em duas tomadas diárias.
Terapia combinada - efavirenz, nevirapina, amprenavir ou nelfinavir
- Um aumento de dose de Kaletra® (lopinavir/ritonavir) para 500 mg/125 mg duas vezes ao dia [2 comprimidos de Kaletra® (lopinavir/ritonavir) 200 mg/50 mg + 1 comprimido de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg] deve ser considerado quando houver coadministração de Kaletra® (lopinavir/ritonavir) com efavirenz, nevirapina, amprenavir ou nelfinavir em pacientes com tratamento antirretroviral prévio, nos quais uma diminuição à susceptibilidade ao lopinavir é clinicamente suspeita (através de histórico de tratamento ou evidência laboratorial), conforme indicação médica.
- Kaletra® (lopinavir/ritonavir) 200 mg/50 mg não deve ser administrado uma única vez ao dia em combinação com efavirenz, nevirapina, amprenavir ou nelfinavir.
- A administração de Kaletra® (lopinavir/ritonavir) 200 mg/50 mg uma única vez ao dia em combinação com indinavir e saquinavir não foi estudada.
Posologia para pacientes pediátricos
Em geral, 91% das crianças entre 06 meses e 11 anos são capazes de deglutir comprimidos pequenos.
No entanto, fica a critério do médico prescritor a escolha pela apresentação que mais se adequa ao paciente pediátrico:
- Solução oral ou comprimidos.
Kaletra® (lopinavir/ritonavir) não deve ser administrado uma vez ao dia em pacientes pediátricos.
- A dose para adultos de Kaletra® (lopinavir/ritonavir) comprimidos revestidos duas vezes ao dia sem administração combinada com efavirenz, nevirapina ou nelfinavir pode ser usada em crianças com 35 kg ou mais, ou com uma Área de Superfície Corporal (ASC) maior ou igual a 1,4 m². Para crianças pesando menos que 35 kg ou com ASC entre 0,6 e 1,4 m² e capazes de engolir comprimidos, seguir tabelas abaixo para definição da dose a ser administrada. Kaletra® (lopinavir/ritonavir) solução oral está disponível para crianças com ASC menor que 0,6 m² e para aquelas incapazes de engolir comprimidos.
Área de Superfície Corporal
A tabela abaixo apresenta o guia para doses pediátricas de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg baseando-se na Área de Superfície Corporal:
|
Guia para doses pediátricas |
|
|
Área de Superfície Corporal (m²) |
Número de comprimidos de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg duas vezes ao dia |
|
≥ 0,6 a < 0,9 |
2 comprimidos (200 mg/50 mg) |
|
≥ 0,9 a < 1,4 |
3 comprimidos (300 mg/75 mg) |
|
≥ 1,4 |
4 comprimidos (400 mg/100 mg) |
A Área de Superfície Corporal (ASC) pode ser calculada a partir da seguinte equação:
- ASC (m²) = √ (Altura (cm) X Peso (kg) / 3600).
Terapia combinada - efavirenz, nevirapina, nelfinavir ou amprenavir
A tabela a seguir contém um guia de doses de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg baseado na Área de Superfície Corporal quando utilizado em combinação com efavirenz, nevirapina, nelfinavir ou amprenavir em crianças.
|
Guia para doses pediátricas com uso juntamente ao efavirenz, nevirapina, nelfinavir ou amprenavir |
|
|
Área de Superfície Corporal (m²) |
Número de comprimidos de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg duas vezes ao dia |
|
≥ 0,6 a < 0,8 |
2 comprimidos (200 mg/50 mg) |
|
≥ 0,8 a < 1,2 |
3 comprimidos (300 mg/75 mg) |
|
≥ 1,2 a < 1,7 |
4 comprimidos (400 mg/100 mg) |
|
≥ 1,7 |
5 comprimidos (500 mg/125 mg) |
Peso
A tabela abaixo apresenta o guia para doses pediátricas de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg baseando-se no peso do paciente:
|
Guia para doses pediátricas |
|
|
Peso (kg) |
Número de comprimidos de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg duas vezes ao dia |
|
7 a < 15 kg |
Não é recomendada a administração de comprimidos. Utilizar solução oral |
|
15 a 25 kg |
2 comprimidos |
|
≥ 25 a 35 kg |
3 comprimidos |
|
> 35 kg |
4 comprimidos* |
*Como alternativa, dois comprimidos de Kaletra® (lopinavir/ritonavir) 200 mg/50 mg podem ser administrados aos pacientes que podem deglutir comprimidos maiores.
Terapia concomitante - efavirenz, nevirapina, nelfinavir ou amprenavir
A tabela a seguir contém um guia de doses de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg baseado no peso do paciente quando utilizado em combinação com efavirenz, nevirapina, nelfinavir ou amprenavir em crianças.
|
Guia para doses pediátricas com uso concomitante de efavirenz, nevirapina, nelfinavir ou amprenavir |
|
|
Peso (kg) |
Número de comprimidos de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg duas vezes ao dia |
|
7 a < 15 kg |
Não é recomendada a administração de comprimidos. Utilizar solução oral |
|
15 a 20 kg |
2 comprimidos |
|
> 20 a 30 kg |
3 comprimidos |
|
> 30 a 45 kg |
4 comprimidos* |
|
> 45 kg |
5 comprimidos |
*Como alternativa, dois comprimidos de Kaletra® (lopinavir/ritonavir) 200 mg/50 mg podem ser administrados aos pacientes que podem deglutir comprimidos maiores.
Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico.
Este medicamento não deve ser partido, aberto ou mastigado.
Superdose: o que acontece se tomar uma dose do Kaletra maior do que a recomendada?
Solução oral / Comprimido revestido
A experiência em casos de superdosagem de Kaletra® (lopinavir/ritonavir) é limitada.
Em caso de superdosagem, cuidados médicos são primordiais. O tratamento envolve medidas médicas de suporte, como monitoramento dos sinais vitais (pulso, pressão, respiração) e observação do paciente.
Não há antídoto específico para estes casos.
Se indicado, pode ser recomendada lavagem gástrica ou indução de vômitos.
Em caso de uso de grande quantidade deste medicamento, procure rapidamente socorro médico e leve a embalagem ou bula do medicamento, se possível. Ligue para 0800 722 6001, se você precisar de mais orientações.
Exclusivo solução oral
Foi relatada superdosagem de Kaletra® (lopinavir/ritonavir) solução oral.
Os seguintes eventos foram reportados em associação com superdosagem não intencionais em recém-nascidos prematuros:
- Bloqueio AV total, cardiomiopatia (doença do músculo cardíaco), acidose láctica (acúmulo de ácido láctico no corpo) e insuficiência renal aguda.
Profissionais de Saúde devem estar cientes que Kaletra® (lopinavir/ritonavir) solução oral é altamente concentrado e contém 42,4% de álcool (v/v) e 15,3% de propilenoglicol (p/v), e portanto, devem prestar especial atenção no cálculo preciso da dose de Kaletra® (lopinavir/ritonavir), a transcrição da ordem de medicação, informação de dispensação e as instruções de dosagem para minimizar o risco de erros de medicação e superdosagem. Isto é especialmente importante para bebês e crianças jovens.
Como Kaletra® (lopinavir/ritonavir) é altamente ligado a proteínas, é pouco provável que a diálise seja benéfica para a remoção significante do fármaco. Entretanto, diálise pode remover tanto álcool como propilenoglicol em casos de superdosagem com Kaletra® (lopinavir/ritonavir).
O que devo fazer quando me esquecer de usar o Kaletra?
Caso esqueça de tomar uma dose de Kaletra® (lopinavir/ritonavir), tome-a tão logo se lembre. Se estiver próximo à dose seguinte, espere e tome a dose no horário previsto. Não duplique a dose seguinte.
Em caso de dúvidas, procure orientação do farmacêutico ou de seu médico, ou cirurgião-dentista.
Quais cuidados devo ter ao usar o Kaletra?
Solução oral / Comprimido revestido
Geral
Atualmente não há dados demonstrando que a terapia com Kaletra® (lopinavir/ritonavir) possa reduzir o risco de transmissão do HIV a outras pessoas pelo contato sexual.
Kaletra® (lopinavir/ritonavir) não deve ser utilizado com certos tipos de medicamentos, pois podem ocorrer efeitos colaterais sérios que podem levar à morte.
Para se prevenir a transmissão do HIV e de outras doenças sexualmente transmissíveis (DSTs), você deve usar corretamente a camisinha nas relações sexuais e apenas agulhas e seringas descartáveis.
Para evitar que o HIV se transmita da mãe para o filho, todas as gestantes devem começar o pré-natal o mais cedo possível e fazer o teste para o HIV.
Diabetes mellitus/hiperglicemia (excesso de glicose no sangue)
Foram relatados aparecimento ou piora de diabetes mellitus preexistente e hiperglicemia em pacientes infectados por HIV. Alguns pacientes necessitaram iniciar ou ajustar as doses de insulina ou de medicamentos para controlar a taxa de açúcar no sangue (hipoglicemiantes orais) para o tratamento destes eventos adversos.
Exclusivo solução oral: Em alguns casos ocorreu cetoacidose diabética. Nos pacientes que descontinuaram a terapia com inibidores de protease, a hiperglicemia persistiu em alguns casos. Deve-se considerar a monitoração da glicemia.
Exclusivo comprimido revestido: Nos pacientes que descontinuaram a terapia com inibidores de protease, a hiperglicemia persistiu em alguns casos. Deve-se considerar a monitoração da glicemia.
Pancreatite
Foi observada pancreatite (inflamação no pâncreas) em pacientes recebendo Kaletra® (lopinavir/ritonavir). Foram observados alguns casos de óbito. A elevação acentuada de triglicérides (gordura no sangue) é um fator de risco para o desenvolvimento de pancreatite. Pacientes com doença avançada pelo HIV podem apresentar risco aumentado de elevação de triglicérides e pancreatite e pacientes com histórico de pancreatite podem apresentar risco aumentado de ter pancreatite novamente.
Insuficiência hepática (falha no funcionamento do fígado)
Kaletra® (lopinavir/ritonavir) é transformado para posterior eliminação principalmente pelo fígado. Portanto, deve-se ter cuidado quando este produto é administrado a pacientes com falha no funcionamento do fígado. Há relatos pós-comercialização do produto de disfunção do fígado, incluindo algumas mortes. De um modo geral, esses eventos ocorreram em pacientes portadores de HIV com a doença avançada, utilizando múltiplos medicamentos concomitantemente e sob tratamento de hepatite (inflamação do fígado) crônica ou cirrose. Não foi estabelecida uma ligação causal com a terapia de Kaletra® (lopinavir/ritonavir).
Foi relatado aumento das enzimas do fígado, com ou sem níveis elevados de bilirrubina em pacientes HIV-1 mono-infectados ou não infectados, após 07 dias do início da terapia de Kaletra® (lopinavir/ritonavir) em conjunto com outros agentes antirretrovirais. Em alguns casos a disfunção hepática foi grave, no entanto, não foi estabelecida uma relação causal definitiva com o tratamento de Kaletra® (lopinavir/ritonavir).
Deve ser considerado um monitoramento frequente de enzimas do fígado nestes pacientes, principalmente nos primeiros meses de tratamento com Kaletra® (lopinavir/ritonavir).
Resistência cruzada
Foram observados vários graus de resistência cruzada entre inibidores de protease, que é a classe de medicamentos à qual pertence o Kaletra® (lopinavir/ritonavir). O efeito do tratamento com Kaletra® (lopinavir/ritonavir) sobre a eficácia de outros inibidores de protease administrados conjuntamente está sendo investigado.
Hemofilia (distúrbio na coagulação do sangue)
Há relatos de sangramento aumentado, incluindo hematomas na pele e hemartrose (sangramento para dentro da articulação) espontâneos em pacientes com hemofilia tipo A e B tratados com inibidores de protease. Em alguns pacientes foi administrado fator VIII adicional. Em mais da metade dos casos relatados o tratamento com inibidores da protease foi mantido ou reiniciado. Não foi estabelecido o mecanismo de ação nem a relação causal entre a terapia com inibidores da protease e estes eventos.
Efeitos no eletrocardiograma
Kaletra® (lopinavir/ritonavir) mostrou causar discreta alteração no eletrocardiograma em alguns pacientes. Kaletra® (lopinavir/ritonavir) deve ser utilizado com cautela em pacientes com insuficiência cardíaca e alterações do ritmo cardíaco.
Elevação de lipídeos
O tratamento com Kaletra® (lopinavir/ritonavir) resultou em aumentos da concentração de colesterol total e triglicérides (gordura no sangue). Devem ser realizados testes de colesterol e triglicérides antes de iniciar a terapia com Kaletra® (lopinavir/ritonavir) e periodicamente durante o tratamento. Veja na seção Interação medicamentosa: quais os efeitos de tomar Kaletra com outros remédios? - Inibidores da HMG-CoA redutase (como pravastatina, fluvastatina, atorvastatina, lovastatina e sinvastatina), informações adicionais sobre interações medicamentosas potenciais de Kaletra® (lopinavir/ritonavir) com esse grupo de medicamentos.
Síndrome da Reconstituição Imunológica
Tal síndrome foi relatada em pacientes infectados por HIV tratados com terapia antirretroviral com diversos medicamentos, incluindo Kaletra® (lopinavir/ritonavir). Durante a fase inicial da terapia antirretroviral combinada, quando o sistema imunológico reage, os pacientes podem desenvolver uma resposta inflamatória a infecções assintomáticas ou a infecções oportunistas latentes (como infecção causada por Mycobacterium avium, citomegalovírus, pneumonia causada por Pneumocystis jiroveci pneumonia, ou tuberculose), podendo necessitar de avaliação e tratamentos adicionais.
Alterações autoimunes [como Doença de Graves (doença que afeta o funcionamento da tireoide), polimiosite (doença inflamatória que afeta os músculos) e Síndrome de Guillain-Barré (doença aguda associada à fraqueza muscular e paralisia)], também foram reportadas durante a fase de reconstituição imunológica, no entanto, o tempo de início é muito variável e podem ocorrer muitos meses após o início do tratamento.
Atenção: o uso incorreto causa resistência do vírus da AIDS e falha no tratamento.
Exclusivo solução oral
Toxicidade em recém-nascidos prematuros
Uma dose segura e eficaz de Kaletra® (lopinavir/ritonavir) solução oral na população de recémnascidos prematuros não foi estabelecida. Kaletra® (lopinavir/ritonavir) solução oral contém o excipiente álcool etílico (42,4% v/v) e propilenoglicol (15,3% p/v). Kaletra® (lopinavir/ritonavir) solução oral não deve ser utilizado por recém-nascidos prematuros no período imediato ao pós-natal devido à possibilidade de toxicidade. O etanol inibe competitivamente o metabolismo do propilenoglicol podendo levar a concentrações elevadas de propilenoglicol. Recém-nascidos prematuros podem apresentar um risco aumentado de reações adversas associadas ao propilenoglicol.
Para todos os medicamentos, incluindo Kaletra® (lopinavir/ritonavir) solução oral, as quantidades totais de álcool e propilenoglicol que são dadas a crianças devem ser levadas em consideração para evitar a toxicidade destes excipientes. As crianças devem ser cuidadosamente monitoradas para aumentos da osmolalidade sérica e creatinina sérica, e para a toxicidade relacionada ao Kaletra® (lopinavir/ritonavir) solução oral, incluindo: hiperosmolalidade, com ou sem acidose láctica (acúmulo de ácido láctico no corpo), toxicidade renal (dos rins), depressão do sistema nervoso central (SNC) [incluindo adormecimento, coma e apneia (ausência de entrada de ar)], convulsões, hipotonia (flacidez muscular), arritmias cardíacas e alterações no ECG e hemólise (destruição dos glóbulos vermelhos).
Casos de risco à vida na pós-comercialização foram relatados como toxicidade cardíaca [incluindo bloqueio AV total, bradicardia (frequência cardíaca diminuída) e cardiomiopatia (doença do músculo cardíaco)], acidose láctica, insuficiência renal aguda, depressão do SNC e complicações respiratórias levando à morte, predominantemente em recém-nascidos prematuros, recebendo Kaletra® (lopinavir/ritonavir) solução oral.
Quais as reações adversas e os efeitos colaterais do Kaletra?
Adultos
O evento adverso mais comum associado ao uso de Kaletra® (lopinavir/ritonavir) foi a diarreia, geralmente de leve a moderada intensidade.
As seguintes reações adversas, de intensidade moderada a grave, com possível ou provável relação com o uso de Kaletra® (lopinavir/ritonavir) foram relatadas por frequência de gravidade:
Reação muito comum (ocorre em mais de 10% dos pacientes que utilizam este medicamento)
- Infecções e infestações: infecção no trato respiratório superior.
- Alterações gastrointestinais: diarreia, náusea.
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Infecções e infestações: infecção no trato respiratório inferior, infecções de pele incluindo celulites (infecção/inflamação da pele), foliculites (infecção dos folículos pilosos causada por bactérias) e furunculose (aparecimento recorrente de furúnculos).
- Alterações no sangue e sistema linfático: anemia, leucopenia (diminuição dos glóbulos brancos) e neutropenia (quantidade de neutrófilos diminuída no sangue), linfadenopatia (aumento dos gânglios linfáticos).
- Alterações no sistema imunológico: hipersensibilidade, incluindo urticária (alergia de pele) e angioedema (inchaço similar à urticária, porém, por baixo da pele).
- Alterações na nutrição e metabolismo: alterações na glicose sanguínea, incluindo diabetes mellitus, hipertrigliceridemia (aumento do triglicérides no sangue), hipercolesterolemia (alto nível de colesterol no sangue), diminuição do peso e diminuição do apetite.
- Alterações psiquiátricas: ansiedade.
- Alterações no sistema nervoso: cefaleia (dor de cabeça), incluindo enxaqueca, neuropatia, incluindo neuropatia periférica (inflamação dos nervos periféricos), vertigem (tontura), insônia.
- Alterações vasculares: hipertensão (pressão alta).
- Alterações gastrointestinais: vômito, doença do refluxo gastroesofágico (DRGE), gastroenterite (infecção estomacal e intestinal) e colite (inflamação no intestino), dor abdominal (superior e inferior), distensão abdominal, pancreatite (inflamação do pâncreas), dispepsia (indigestão), hemorroidas e flatulência (gases intestinais).
- Alterações hepatobiliares: hepatite (inflamação no fígado), incluindo aumento das enzimas do fígado aspartato aminotransferase (AST), alanina aminotransferase (ALT) e gama glutamil transferase (GGT).
- Alterações na pele e tecido subcutâneo: rash, incluindo rash maculopapular (vermelhidão na pele), dermatite/rash, incluindo eczema (pele áspera) e dermatite seborreica (doença de pele que ataca principalmente o couro cabeludo), suores noturnos, prurido (coceira).
- Alterações no tecido conectivo e musculoesquelético: mialgia (dor nos músculos), dor musculoesquelética, incluindo artralgia (dor nas articulações) e dor nas costas, alterações musculares como fraqueza e espasmos.
- Alterações renais e urinárias: insuficiência renal (mau funcionamento dos rins).
- Alterações no sistema reprodutivo e mamas: disfunção erétil, alterações menstruais como amenorreia (ausência de menstruação), menorragia (menstruação extremamente abundante ou prolongada).
- Alterações gerais e nas condições de administração: fadiga, incluindo astenia (fraqueza).
Reação incomum (ocorre entre 0,1% e 1% dos pacientes que utilizam este medicamento)
- Alterações no sistema imunológico: síndrome da reconstituição imune.
- Alterações endócrinas: hipogonadismo (secreção inadequada de testosterona pelos testículos).
- Alterações na nutrição e metabolismo: aumento de peso, aumento de apetite, acidose láctica.
- Alterações psiquiátricas: sonhos anormais, diminuição da libido.
- Alterações no sistema nervoso: evento cerebrovascular, convulsão, ageusia (ausência ou diminuição do paladar), tremor.
- Alterações nos olhos: deficiência visual.
- Alterações no ouvido e labirinto: tinido, tontura.
- Alterações cardíacas: aterosclerose (formação de placas na parede das artérias do coração), como infarto do miocárdio, bloqueio atrioventricular, insuficiência da válvula tricúspide.
- Alterações vasculares: trombose venosa profunda.
- Alterações gastrointestinais: hemorragia gastrointestinal, incluindo hemorragia retal, úlcera gastrointestinal, duodenite (inflamação do duodeno) e gastrite, estomatite (inflamação da boca ou gengivas) e úlceras na boca, incontinência fecal, constipação (prisão de ventre), boca seca.
- Alterações hepatobiliares: esteatose hepática (acúmulo de gordura no fígado), hepatomegalia (tamanho do fígado aumentado), colangite (inflamação das vias biliares).
- Alterações na pele e tecido subcutâneo: alopecia (queda de cabelo), capilarite (inflamação dos vasos capilares), vasculite (inflamação nos vasos sanguíneos).
- Alterações no tecido conectivo e musculoesquelético: rabdomiólise (destruição muscular), osteonecrose (necrose do osso).
- Alterações renais e urinárias: nefrite (inflamação nos rins), hematúria (sangue na urina).
Pacientes pediátricos
Em crianças com 02 anos de idade ou mais, o perfil de eventos adversos vistos durante o estudo clínico em pacientes pediátricos foi similar àqueles apresentados pelos pacientes adultos.
Reação comum (ocorre entre 1% e 10% dos pacientes que utilizam este medicamento)
- Infecção por vírus, disgeusia (distorção ou diminuição do senso do paladar), constipação (prisão de ventre), vômito, pancreatite (inflamação do pâncreas), hepatomegalia (tamanho do fígado aumentado), rash, pele seca e febre.
Experiência pós-comercialização
- Alterações hepatobiliares: hepatite (inflamação do fígado) foi relatada em pacientes que utilizaram Kaletra® (lopinavir/ritonavir).
- Alterações na pele e tecido subcutâneo: necrólise epidérmica tóxica, Síndrome de Stevens-Johnson e eritema multiforme foram relatados.
- Alterações cardíacas: bradiarritmia (frequência cardíaca lenta) foi relatada.
- Alterações renais e urinárias: nefrolitíase (pedras nos rins).
Informe ao seu médico, cirurgião-dentista ou farmacêutico o aparecimento de reações indesejáveis pelo uso do medicamento. Informe também à empresa através do seu serviço de atendimento.
População Especial do Kaletra
Uso em crianças
Os perfis de segurança e ação do medicamento não foram estabelecidos para pacientes com menos de 06 meses de idade.
Em pacientes infectados pelo HIV com idades entre 06 meses e 18 anos, o perfil de reações adversas observado durante um estudo clínico foi semelhante ao observado em pacientes adultos. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma vez ao dia na população pediátrica.
Exclusivo solução oral: Para uso de Kaletra® (lopinavir/ritonavir) solução oral na pediatria, veja seções: “Quais cuidados devo ter ao usar o Kaletra?”; “Como usar o Kaletra?” e “Superdose: o que acontece se tomar uma dose do Kaletra maior do que a recomendada?”.
Uso em idosos
Os estudos clínicos com Kaletra® (lopinavir/ritonavir) não incluíram um número suficiente de indivíduos com mais de 65 anos para determinar se estes respondem diferentemente ao tratamento em relação a indivíduos mais jovens. Em geral, deve-se ter cuidado na administração e monitoramento de Kaletra® (lopinavir/ritonavir) em pacientes idosos devido à maior frequência de função hepática, renal ou cardíaca diminuídas e de doenças ou outros tratamentos medicamentosos concomitantes.
Gravidez, fertilidade e reprodução
Kaletra® (lopinavir/ritonavir) deve ser usado durante a gravidez somente quando, na opinião do médico, os benefícios potenciais claramente justificarem os possíveis riscos.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso na lactação
Por causa do potencial de transmissão do HIV e possíveis reações adversas de Kaletra® (lopinavir/ritonavir), as mães devem ser instruídas a não amamentar enquanto estiverem recebendo Kaletra® (lopinavir/ritonavir). É desconhecido se o lopinavir é excretado no leite humano.
Uso em pacientes com insuficiência renal
Lopinavir não foi estudado em pacientes com insuficiência renal (dos rins), entretanto, não são esperadas alterações nesta população de pacientes.
Uso em pacientes com insuficiência hepática
Doses múltiplas de Kaletra® (lopinavir/ritonavir) em pacientes coportadores de HIV e hepatite C (HCV) com insuficiência hepática (do fígado) leve a moderada resultaram em aumento de lopinavir no sangue quando comparados com pacientes portadores de HIV com função hepática normal. Kaletra® (lopinavir/ritonavir) não foi estudado em pacientes com insuficiência hepática grave.
Interação medicamentosa: quais os efeitos de tomar Kaletra com outros remédios?
Solução oral / Comprimido revestido
| Agentes antigotosos | Interações medicamentosas fatais ou de risco à vida foram reportadas em pacientes tratados com colchicina e inibidores fortes de CYP3A4, como o ritonavir |
| Antimicobacterianos | Rifampicina não deve ser utilizada concomitantemente a Kaletra® (lopinavir/ritonavir) por causa da grande redução que ocorre nas concentrações de lopinavir, o que pode diminuir significativamente seu efeito terapêutico. A coadministração de bedaquilina e Kaletra® (lopinavir/ritonavir) pode aumentar o risco de reações adversas relacionadas à bedaquilina. A bedaquilina deve ser usada cautelosamente com Kaletra® (lopinavir/ritonavir), ou seja, somente quando na opinião do médico o benefício da coadministração for superior ao risco. A coadministração de delamanide com um potente inibidor da CYP3A (lopinavir/ritonavir) pode aumentar ligeiramente a exposição ao metabólito delamanide, que tem sido associada com o prolongamento do intervalo QTc. Portanto, se a coadministração de delamanide com lopinavir/ritonavir é considerada necessária, é recomendada a monitorização frequente por eletrocardiograma (ECG),durante todo o período de tratamento com delamanide |
| Antipsicóticos | Deve-se ter cautela no uso concomitante de Kaletra® (lopinavir/ritonavir) e quetiapina. Devido à inibição da enzima CYP3A por lopinavir/ritonavir, espera-se um aumento das concentrações de quetiapina, podendo levar a efeitos tóxicos relacionados a este antipsicótico |
| Corticosteroides | O uso concomitante de Kaletra® (lopinavir/ritonavir) e fluticasona (inalatória, injetável ou intranasal), budesonida, triancinolona ou outro glicocorticoide que é metabolizado pela enzima CYP3A4, não é recomendado a menos que os benefícios potenciais do tratamento sobreponham os riscos dos efeitos sistêmicos dos corticosteroides, incluindo Síndrome de Cushing (aumento de cortisol no sangue) e supressão adrenal (diminuição da atividade da glândula adrenal). O uso concomitante de propionato de fluticasona e Kaletra® (lopinavir/ritonavir) pode aumentar a concentração de propionato de fluticasona e reduzir os níveis sanguíneos de cortisol. Efeitos dos corticosteroides, incluindo Síndrome de Cushing e supressão adrenal, foram reportados quando houve a administração combinada a propionato de fluticasona, budesonida ou triancinolona injetável |
| Inibidores de PDE5 | A coadministração de Kaletra® (lopinavir/ritonavir) com avanafil não é recomendada. Deve-se ter cautela ao prescrever sildenafila, tadalafila e vardenafila para o tratamento de disfunção erétil em pacientes recebendo Kaletra® (lopinavir/ritonavir). É esperado que a coadministração de Kaletra® (lopinavir/ritonavir) e estas substâncias aumentem a concentração destes agentes, o que pode levar ao aumento de reações adversas, como hipotensão e ereção prolongada. O uso concomitante de sildenafila e Kaletra® (lopinavir/ritonavir) é contraindicado em casos de hipertensão (pressão alta) arterial pulmonar |
| Produtos fitoterápicos | Erva-de-São-João (Hypericum perforatum) pode reduzir substancialmente a concentração de lopinavir e de outros inibidores de protease e, portanto, o uso concomitante não é indicado. Esta associação pode resultar em perda do efeito terapêutico e desenvolvimento de resistência ao lopinavir ou à classe de inibidores de protease |
| Inibidores da HMG-CoA redutase | O uso concomitante de lovastatina ou sinvastatina e Kaletra® (lopinavir/ritonavir) é contraindicado. Deve-se ter cautela ao utilizar inibidores de protease, como Kaletra® (lopinavir/ritonavir), juntamente a rosuvastatina ou outros inibidores de HMG-CoA redutase, tais como a atorvastatina, já que esta combinação pode aumentar o potencial para reações graves, como a miopatia, incluindo rabdomiólise (destruição muscular) |
| Tipranavir | A administração concomitante de Kaletra® (lopinavir/ritonavir) e tipranavir com baixa dose de ritonavir não é recomendada |
Kaletra® (lopinavir/ritonavir) é metabolizado (transformado) no fígado por uma enzima chamada CYP3A. Quando outros medicamentos são também metabolizados da mesma forma, podem ocorrer efeitos colaterais decorrentes do aumento ou diminuição dos níveis do fármaco.
Exemplos de medicamentos metabolizados pela enzima CYP3A:
- Bloqueadores de canal de cálcio derivados da di-hidropiridina, inibidores da HMG-CoA redutase, imunossupressores e inibidores da enzima PDE5 (sildenafila, tadalafila e vardenafila).
Os exemplos mencionados são um guia e não são considerados uma lista completa de todos os possíveis medicamentos que podem interagir com o Kaletra® (lopinavir/ritonavir). O seu médico deve consultar referências apropriadas para informações mais abrangentes.
Medicamentos com importante potencial de interação
- Antivirais de ação direta (DAA) para o tratamento da Hepatite C - boceprevir (inibidor da protease do Vírus da Hepatite C - HCV): a administração concomitante de boceprevir e Kaletra® (lopinavir/ritonavir) resultou na diminuição de boceprevir e lopinavir no sangue. A coadministração de boceprevir e Kaletra® (lopinavir/ritonavir) não é recomendada.
- Atorvastatina: quando a administração em conjunto com atorvastatina estiver indicada, deve-se utilizar a menor dose possível. As interações com pravastatina e fluvastatina não são esperadas. Se houver indicação de tratamento combinado de Kaletra® (lopinavir/ritonavir) com um inibidor da HMGCoA redutase, recomenda-se utilizar pravastatina ou fluvastatina.
- Bedaquilina: a administração de bedaquilina com Kaletra® (lopinavir/ritonavir) pode aumentar a concentração de bedaquilina no sangue. A bedaquilina deve ser usada cautelosamente com Kaletra® (lopinavir/ritonavir), ou seja, somente quando na opinião do médico o benefício da coadministração for superior ao risco.
- Lovastatina e sinvastatina: os inibidores da HMG-CoA redutase, medicamentos que atuam na redução do colesterol, tais como a lovastatina e sinvastatina, podem apresentar um aumento acentuado de suas concentrações plasmáticas quando administrados juntamente a Kaletra® (lopinavir/ritonavir). Considerando-se que as concentrações aumentadas de inibidores da HMG-CoA redutase podem causar alterações nos músculos, incluindo a destruição muscular, a combinação desses medicamentos com Kaletra® (lopinavir/ritonavir) é contraindicada.
- Contraceptivos orais e adesivos: considerando que os níveis de etinilestradiol podem ser reduzidos, deve-se utilizar um método contraceptivo alternativo ou adicional quando houver indicação de uso de Kaletra® (lopinavir/ritonavir) juntamente a contraceptivos orais e adesivos a base de estrógeno.
- Propionato de fluticasona (inalatória, injetável ou intranasal), budesonida e triancinolona: o uso concomitante de propionato de fluticasona ou outro glicocorticoide que é metabolizado pela CYP3A4, como a budesonida, e Kaletra® (lopinavir/ritonavir) não é recomendado, a menos que na opinião do médico os benefícios potenciais do tratamento sobreponham os riscos dos efeitos sistêmicos dos corticoides, incluindo Síndrome de Cushing (aumento de cortisol no sangue) e supressão adrenal (diminuição da atividade da glândula adrenal). O médico deve considerar medicamentos alternativos ao propionato de fluticasona, budesonida ou triancinolona injetável, particularmente quando o uso for prolongado.
- Fosamprenavir: a administração conjunta de Kaletra® (lopinavir/ritonavir) e fosamprenavir diminui a concentração de amprenavir e lopinavir.
- Rifampicina: não deve ser utilizada concomitantemente a Kaletra® (lopinavir/ritonavir) por causa da grande redução que ocorre nas concentrações de lopinavir. O uso de rifampicina com Kaletra® (lopinavir/ritonavir) pode levar a uma perda da resposta virológica e possivelmente resistência ao Kaletra® (lopinavir/ritonavir), à classe dos inibidores de protease ou a outros agentes antirretrovirais coadministrados. Se a coadministração for considerada, Kaletra® (lopinavir/ritonavir) deve ser iniciado com doses padronizadas por aproximadamente 10 dias antes da adição da rifampicina. Somente então a dose de Kaletra® (lopinavir/ritonavir) deve ser titulada. A função hepática deve ser monitorada com atenção.
- Sildenafila: o uso de sildenafila em combinação a Kaletra® (lopinavir/ritonavir) é contraindicado em pacientes com hipertensão arterial pulmonar.
- Produtos fitoterápicos: erva-de-São-João (Hypericum perforatum) pode reduzir substancialmente a concentração de Kaletra® (lopinavir/ritonavir) e, portanto, esta combinação é contraindicada.
- Voriconazol: a combinação de Kaletra® (lopinavir/ritonavir) e voriconazol deve ser evitada, a não ser que o risco-benefício justifique o uso de voriconazol.
Medicamentos com recomendação de alteração ou monitoramento da dose
- Agentes antigotosos: é esperado um aumento nas concentrações de colchicina quando coadministrada com Kaletra® (lopinavir/ritonavir). Interações medicamentosas fatais e de risco à vida têm sido reportadas em pacientes tratados com colchicina e ritonavir. Remeter à bula de colchicina para informações de prescrição.
- Agentes anticancerígenos (abemaciclibe, apalutamida, dasatinibe, encorafenibe, ibrutinibe, ivosidenibe, neratinibe, nilotinibe, venetoclax, vincristina e vimblastina): podem ter suas concentrações aumentadas quando administrados com Kaletra® (lopinavir/ritonavir), resultando em aumento dos eventos adversos, alguns dos quais podendo ser graves. A coadministração de venetoclax ou ibrutinibe com Kaletra® (lopinavir/ritonavir) pode aumentar potencialmente a exposição à venetoclax ou ibrutinibe, resultando em um sério risco de Síndrome da Lise Tumoral. A coadministração de encorafenibe ou ivosidenibe com Kaletra® (lopinavir/ritonavir) pode aumentar a exposição de encorafenibe ou ivosidenibe, aumentando potencialmente o risco de eventos adversos graves, como o prolongamento do intervalo QT. Para venetoclax, encorafenibe, ibrutinibe, ivosidenibe, nilotinibe e dasatinibe, consultar suas informações de prescrição para instruções de dose. A coadministração de apalutamida com Kaletra® (lopinavir/ritonavir) é contraindicada, uma vez que apalutamida pode diminuir a exposição de Kaletra® (lopinavir/ritonavir) com potencial perda de resposta. Adicionalmente, a administração concomitante de apalutamida e Kaletra® (lopinavir/ritonavir) pode aumentar a exposição da apalutamida, resultando em um aumento potencial para eventos adversos, incluindo convulsões.
- Agentes vasodilatadores: a coadministração de bosentana e Kaletra® (lopinavir/ritonavir) aumenta a concentração de bosentana no sangue. Remeter à bula de bosentana para informações de prescrição.
- Antagonistas do receptor do hormônio liberador de gonadotrofina (GnRH): a coadministração de elagolix com Kaletra® (lopinavir/ritonavir) pode aumentar a exposição ao elagolix.. Eventos adversos graves conhecidos de elagolix incluem ideação suicida e elevação de transaminases hepáticas. Adicionalmente, elagolix é um indutor fraco/moderado de CYP3A, o que pode aumentar a exposição ao Kaletra® (lopinavir/ritonavir). Consulte a bula de elagolix para informações sobre dosagem com inibidores fortes de CYP-3A4.
- Antiarrítmicos: as concentrações de amiodarona, bepridila, dronedarona, lidocaína e quinidina podem ser aumentadas quando administradas juntamente a Kaletra® (lopinavir/ritonavir). Recomenda-se cuidado.
- Digoxina: a coadministração de ritonavir e digoxina resulta em um aumento significativo dos níveis de digoxina. Atenção especial deve ser dada quando houver administração combinada destas substâncias, com monitoramento dos níveis sanguíneos de digoxina.
- Anticonvulsivantes: carbamazepina, fenobarbital e fenitoína podem reduzir as concentrações de lopinavir. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com fenobarbital, fenitoína ou carbamazepina. A administração combinada de Kaletra® (lopinavir/ritonavir) e fenitoína pode resultar em diminuição moderada nas concentrações de fenitoína.
- Antivirais de ação direta (DAA) para o tratamento da Hepatite C:
- Glecaprevir / pibrentasvir: a administração concomitante de glecaprevir/pibrentasvir e lopinavir/ritonavir não é recomendada devido ao alto risco de elevação da ALT (enzima do fígado) associada com elevada exposição ao glecaprevir.
- Ombitasvir / veruprevir / ritonavir / dasabuvir: as concentrações de ombitasvir, veruprevir e ritonavir podem ser aumentadas quando coadministradas com Kaletra® (lopinavir/ritonavir). Portanto, a administração concomitante não é recomendada.
- Simeprevir: o uso concomitante de Kaletra® (lopinavir/ritonavir) e simeprevir pode resultar em um aumento da concentração de simeprevir no sangue. Não é recomendado coadministrar Kaletra® (lopinavir/ritonavir) e simeprevir.
- Sofosbuvir / velpatasvir / voxilaprevir: a administração concomitante de sofosbuvir/velpatasvir/voxilaprevir e lopinavir/ritonavir não é recomendada devido ao potencial aumento de toxicidade, que pode ter impacto negativo no uso.
- Telaprevir (inibidor da protease do HCV): a coadministração de telaprevir e Kaletra® (lopinavir/ritonavir) resultou em uma redução da concentração de telaprevir, enquanto lopinavir não foi afetado.
- Atovaquona: pode ocorrer diminuição dos níveis terapêuticos da atovaquona, podendo ser requerida doses maiores desta substância quando da administração concomitante a Kaletra® (lopinavir/ritonavir).
- Claritromicina: para pacientes com insuficiência renal ou hepática (falha no rim ou fígado), deve ser considerada a redução na dose de claritromicina.
- Delamanide: em um estudo de interação medicamentosa com voluntários saudáveis administrou-se delamanide 100 mg duas vezes ao dia e lopinavir/ritonavir 400 mg/100 mg duas vezes ao dia por 14 dias. As exposições de delamanide e um metabólito do delamanide, DM-6705, aumentaram ligeiramente. Caso a coadministração de delamanide com lopinavir/ritonavir for considerada necessária, devido ao risco de prolongamento do QTc associada ao DM-6705, recomenda-se a monitorização frequente por ECG durante todo o período de tratamento com delamanide.
- Nevirapina e efavirenz: a nevirapina pode reduzir os níveis de lopinavir no sangue. Para pacientes que fizeram uso prévio de inibidores de protease ou com perda significante de sensibilidade ao lopinavir, pode ser considerado um aumento de dose do Kaletra® (lopinavir/ritonavir) quando administrado em combinação a nevirapina ou efavirenz. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com nevirapina ou efavirenz.
- Fentanila: é esperado um aumento da concentração plasmática de fentanila quando administrada com Kaletra® (lopinavir/ritonavir). Monitoramento cuidadoso da terapia e eventos adversos (incluindo depressão respiratória) pelo médico é recomendado quando fentanila é administrada concomitantemente a Kaletra® (lopinavir/ritonavir).
- Imunossupressores: as concentrações de ciclosporina, tacrolimo e sirolimo (rapamicina) podem aumentar quando administradas juntamente a Kaletra® (lopinavir/ritonavir). Recomenda-se cautela na coadministração destas drogas.
- Indinavir: espera-se que Kaletra® (lopinavir/ritonavir) aumente as concentrações de indinavir. Pode ser necessário diminuir a dose de indinavir durante a administração com Kaletra® (lopinavir/ritonavir). Kaletra® (lopinavir/ritonavir) administrado uma única vez ao dia não foi estudado em combinação com indinavir.
- Cetoconazol/itraconazol: o cetoconazol e o itraconazol podem apresentar concentrações sanguíneas aumentadas pelo Kaletra® (lopinavir/ritonavir).
- Lamotrigina e valproato: a administração concomitante de Kaletra® (lopinavir/ritonavir) e qualquer um destes medicamentos foi associada com uma redução do anticonvulsivante. Utilizar com cuidado. Um aumento de dose do anticonvulsivante pode ser necessário quando coadministrado com Kaletra® (lopinavir/ritonavir) e um monitoramento da concentração terapêutica do anticonvulsivante pode ser indicado pelo médico, particularmente durante o ajuste de dose.
- Maraviroque (antagonistas de CCR5): a administração concomitante de maraviroque com Kaletra® (lopinavir/ritonavir) aumenta os níveis plasmáticos de maraviroque. A dose de maraviroque deve ser diminuída durante a coadministração com Kaletra® (lopinavir/ritonavir). Para mais detalhes, veja as informações de prescrição de maraviroque.
- Metadona: Kaletra® (lopinavir/ritonavir) apresentou redução das concentrações plasmáticas da metadona e, por isso, recomenda-se cautela na coadministração destas drogas.
- Nelfinavir: espera-se que Kaletra® (lopinavir/ritonavir) aumente as concentrações de nelfinavir e que esta combinação resulte em uma diminuição das concentrações de lopinavir. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com nelfinavir.
- Quetiapina: devido à inibição da enzima CYP3A por lopinavir/ritonavir, espera-se um aumento das concentrações de quetiapina. Para instruções de dose de quetiapina, consultar suas informações de prescrição.
- Rifabutina: recomenda-se uma redução da dose da rifabutina quando houver indicação de uso concomitante a Kaletra® (lopinavir/ritonavir). Poderá ser necessária posterior redução da dose de rifabutina.
- Rivaroxabana: a coadministração de rivaroxabana e Kaletra® (lopinavir/ritonavir) pode aumentar a exposição de rivaroxabana, o que pode aumentar o risco de sangramento.
- Saquinavir: espera-se que Kaletra® (lopinavir/ritonavir) aumente as concentrações de saquinavir. Pode ser necessária uma diminuição da dose de saquinavir quando administrado em combinação a Kaletra® (lopinavir/ritonavir). Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com saquinavir.
- Inibidores de PDE5: recomenda-se cautela no uso de sildenafila, tadalafila e vardenafila para o tratamento de disfunção erétil em pacientes recebendo Kaletra® (lopinavir/ritonavir). É esperado que essa associação aumente substancialmente as concentrações destas substâncias no sangue, o que pode levar ao aumento de reações adversas, como hipotensão (pressão baixa) e ereção persistente.
- Avanafil: a coadministração de Kaletra® (lopinavir/ritonavir) com avanafil pode resultar em um grande aumento na exposição à avanafil, logo, essa coadministração não é recomendada.
- Sildenafila: a sildenafila para tratamento da disfunção erétil deve ser utilizada com cautela em doses reduzidas de 25 mg a cada 48 horas com monitoramento dos eventos adversos.
- Tadalafila: use tadalafila com atenção em doses reduzidas de no máximo 10 mg a cada 72 horas com monitoramento intensivo dos eventos adversos. Quando tadalafila é administrada para o tratamento de hipertensão arterial pulmonar em pacientes recebendo Kaletra® (lopinavir/ritonavir), remeter à bula de tadalafila para informações de prescrição.
- Vardenafila: use vardenafila com atenção em doses reduzidas de no máximo 2,5 mg a cada 72 horas com monitoramento intensivo dos eventos adversos.
- Tenofovir: um estudo mostrou que Kaletra® (lopinavir/ritonavir) aumenta a concentração de tenofovir. Pacientes utilizando esta combinação devem ser monitorados em relação aos eventos adversos associados ao tenofovir.
- Trazodona: o uso concomitante de ritonavir e trazodona pode aumentar a concentração de trazodona. Eventos adversos como náuseas, vertigens, hipotensão (pressão baixa) e desmaio foram observados. A combinação deve ser usada com atenção e uma dose menor de trazodona pode ser considerada.
- Varfarina: a concentração de varfarina pode ser afetada quando administrada em combinação a Kaletra® (lopinavir/ritonavir). Recomenda-se cautela na coadministração destas drogas.
Outras Interações Medicamentosas
- Bupropiona: a administração conjunta de Kaletra® (lopinavir/ritonavir) e bupropiona diminui a concentração sanguínea da bupropiona.
- Delavirdina: a delavirdina tem o potencial de aumentar as concentrações plasmáticas de lopinavir.
- Bloqueadores de canal de cálcio: derivados da di-hidropiridina (felodipino, nifedipino, nicardipino) podem ter as suas concentrações aumentadas quando administrados juntamente a Kaletra® (lopinavir/ritonavir).
- Dexametasona: pode reduzir as concentrações de lopinavir.
- Inibidores de quinase: a coadministração de fostamatinibe com Kaletra® (lopinavir/ritonavir) pode aumentar a exposição do fostamatinibe, resultando em eventos adversos relacionados à dosagem, como hepatotoxicidade e neutropenia.
- Ritonavir: quando Kaletra® (lopinavir/ritonavir) foi coadministrado a mais 100 mg de ritonavir duas vezes ao dia, os níveis de lopinavir aumentaram no sangue.
- Etravirina: o uso concomitante de Kaletra® (lopinavir/ritonavir) com etravirina causa uma diminuição na concentração de etravirina no sangue, porém, não é necessário ajuste de dose pelo médico. Remeter à bula de etravirina para informações de prescrição.
- Rilpivirina: o uso concomitante de Kaletra® (lopinavir/ritonavir) com rilpivirina causa um aumento na concentração de rilpivirina no sangue, porém, não é necessário ajuste de dose pelo médico. Remeter à bula de rilpivirina para informações de prescrição.
- Inibidores Nucleosídicos da Transcriptase Reversa (ITRNs): aumento dos níveis séricos de creatina fosfoquinase (CPK) no sangue (exame laboratorial), dor muscular, inflamação muscular e raramente rabdomiólise (destruição muscular) foram relatados com inibidores de protease, particularmente em combinação com ITRNs.
- Zidovudina e abacavir: Kaletra® (lopinavir/ritonavir) apresenta potencial para reduzir as concentrações sanguíneas de zidovudina e abacavir.
Interação Medicamentosa com significado clínico não esperado
Estudos de interações medicamentosas revelaram que não há interação clinicamente significativa com Kaletra® (lopinavir/ritonavir) e desipramina, omeprazol ou ranitidina.
Estudos não demonstraram interação clinicamente significativa entre Kaletra® (lopinavir/ritonavir) e raltegravir.
Não são esperadas interações medicamentosas significativas entre Kaletra® (lopinavir/ritonavir) e fluvastatina, dapsona, trimetoprima/sulfametoxazol, azitromicina ou fluconazol em pacientes com funções normais de rim e fígado.
Para qualquer interação com outros medicamentos mencionada nesta bula, alterações na dose recomendada de Kaletra® (lopinavir/ritonavir) somente deverão ser efetuadas pelo médico.
- Estavudina e lamivudina: nenhuma alteração na farmacocinética do lopinavir foi observada quando Kaletra® (lopinavir/ritonavir) foi administrado isoladamente ou em combinação com estavudina ou lamivudina.
- Fármacos redutores de acidez gástrica: Kaletra® (lopinavir/ritonavir) pode ser utilizado em combinação com fármacos redutores de acidez gástrica (omeprazol e ranitidina) sem a necessidade de ajuste de dose.
Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento.
Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para sua saúde.
Exclusivo solução oral
Medicamentos com importante potencial de interação
- Lomitapida: a lomitapida é um substrato sensível para o metabolismo do CYP3A4. Os inibidores do CYP3A4 aumentam a exposição da lomitapida, em uso com fortes inibidores, aumentando a exposição em aproximadamente 27 vezes. O uso concomitante de inibidores moderados ou fortes do CYP3A4 com lomitapida é contraindicado.
- Dissulfiram/metronidazol: Kaletra® (lopinavir/ritonavir) solução oral contém álcool, que pode produzir reações típicas do dissulfiram quando houver a coadministração com dissulfiram ou outros medicamentos que produzem as mesmas reações, como metronidazol.
Medicamentos com recomendação de alteração ou monitoramento da dose
- Amprenavir: espera-se que Kaletra® (lopinavir/ritonavir) aumente as concentrações de amprenavir no sangue. A administração em combinação de Kaletra® (lopinavir/ritonavir) e amprenavir resulta em redução nas concentrações de lopinavir no sangue. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com amprenavir.
Outras Interações Medicamentosas
- Inibidores Nucleosídicos da Transcriptase Reversa (ITRNs):
- Didanosina: é recomendado que a didanosina seja administrada com estômago vazio, portanto, a didanosina deve ser administrada uma ou duas horas antes da administração de Kaletra® (lopinavir/ritonavir) solução oral.
Exclusivo comprimido revestido
Medicamentos com importante potencial de interação
- Lomitapida: a exposição da lomitapida é aumentada em uso com fortes inibidores da CYP3A4. O uso concomitante de inibidores moderados ou fortes do CYP3A4 com lomitapida é contraindicado.
Medicamentos com recomendação de alteração ou monitoramento da dose
- Amprenavir: espera-se que Kaletra® (lopinavir/ritonavir) aumente as concentrações de amprenavir no sangue. A administração em combinação de Kaletra® (lopinavir/ritonavir) e amprenavir resultam em redução nas concentrações de lopinavir no sangue. Pode ser necessário um aumento de dose de Kaletra® (lopinavir/ritonavir) durante a coadministração com amprenavir, particularmente em pacientes com larga experiência de uso de inibidores de protease ou com evidências de perda significante de sensibilidade para o lopinavir. Kaletra® (lopinavir/ritonavir) não deve ser administrado uma única vez ao dia em combinação com amprenavir.
Outras Interações Medicamentosas
- Inibidores Nucleosídicos da Transcriptase Reversa (ITRNs):
- Didanosina: é recomendado que a didanosina seja administrada com estômago vazio, portanto, a didanosina pode ser administrada com Kaletra® (lopinavir/ritonavir) comprimidos revestidos sem alimentos.
Apresentações do Kaletra
Solução oral 80 mg/mL + 20 mg/mL
Embalagem com 1 frasco contendo 160 mL de solução oral. Inclui copo medida para administração oral.
Uso oral.
Uso adulto e pediátrico acima de 06 meses de idade.
Comprimidos revestidos
- 200 mg/50 mg (lopinavir/ritonavir) - embalagem com 120 comprimidos revestidos;
- 100 mg/25 mg (lopinavir/ritonavir) - embalagem com 60 comprimidos revestidos.
Uso oral.
Uso adulto e pediátrico acima de 06 meses de idade (para crianças capazes de deglutir comprimidos).
Qual a ação da substância do Kaletra?
Resultados de Eficácia
Pacientes Sem Terapia Antirretroviral Prévia
Estudo M98-863: Lopinavir + Ritonavir cápsulas duas vezes ao dia + estavudina + lamivudina em comparação a nelfinavir três vezes ao dia + Estavudina + Lamivudina
O Estudo M98-863 foi um ensaio randomizado, duplo-cego, multicêntrico, comparando o tratamento com Lopinavir + Ritonavir cápsulas (400/100 mg duas vezes ao dia) mais estavudina e lamivudina versus nelfinavir (750 mg três vezes ao dia) mais estavudina e lamivudina em 653 pacientes sem tratamento antirretroviral prévio (naive).
Os pacientes tinham uma média de idade de 38 anos (faixa: 19 a 84), 57% eram caucasianos e 80% eram do sexo masculino. A contagem celular média basal de CD4 foi de 259 células/mm³ (faixa: 2 a 949 células/mm³) e a concentração plasmática basal média de RNA HIV-1 foi de 4,9 log10 cópias/mL (faixa: 2,6 a 6,8 log10 cópias/mL).
A resposta ao tratamento e os resultados do tratamento randomizado estão presentes na figura a seguir e na tabela a seguir, respectivamente:

* Proporção de pacientes que, a cada momento na escala de tempo, atingiram e mantiveram uma concentração plasmática de RNA do HIV <400 cópias/mL, e que estão sob sua medição original do estudo e que não experimentaram um novo evento CDC Classe C.
** BID - Bis in die ou duas vezes ao dia.
|
Resultados do Tratamento Randomizado até a Semana 48 (Estudo 863) |
||
|
Resultado |
Lopinavir + Ritonavir + d4T + 3TC (N=326) |
Nelfinavir + d4T + 3TC (N=327) |
|
Respondedores*1 |
75% |
62% |
|
Falha virológica2 |
9% |
25% |
|
Rebote2 |
7% | 15% |
|
Nunca suprimiu até a Semana 482 |
2% | 9% |
|
Morte |
2% | 1% |
|
Descontinuou por evento adverso |
4% | 4% |
|
Descontinuou por outras razões3 |
10% | 8% |
* Corresponde às taxas na Semana 48 da Figura acima.
1 Pacientes atingiram e mantiveram RNA HIV confirmado de <400 cópias/mL até a Semana 48.
2 Inclui as situações confirmadas de rebotes virais e falhas para atingir <400 cópias/mL, até a Semana 48.
3 Inclui perda de acompanhamento, retirada do paciente, não-aderência, violação do protocolo e outras razões.
Descontinuação global até a Semana 48, incluindo pacientes que descontinuaram após a ocorrência de falha virológica, foi de 17% no grupo com Lopinavir + Ritonavir e de 24% no grupo com Nelfinavir.
Ao longo de 48 semanas de tratamento, houve uma proporção estatística e significativamente maior de pacientes no braço com Lopinavir + Ritonavir , em comparação ao braço com Nelfinavir, que apresentaram níveis plasmáticos de RNA HIV menores que 400 cópias/mL (75% versus 62%, respectivamente) e RNA HIV <50 cópias/mL (67% versus 52%, respectivamente).
A resposta ao tratamento pelos subgrupos de nível basal de RNA HIV é apresentada na tabela a seguir:
 1 Pacientes atingiram e mantiveram concentração plasmática confirmada do RNA do HIV <400 cópias/mL até a Semana 48.
1 Pacientes atingiram e mantiveram concentração plasmática confirmada do RNA do HIV <400 cópias/mL até a Semana 48.
2 Pacientes atingiram RNA HIV <50 cópias/mL até a Semana 48.
Ao longo de 48 semanas de tratamento, o aumento médio desde a linha basal na contagem de células CD4 foi de 207 células/mm³ para o braço com Lopinavir + Ritonavir e de 195 células/mm³ para o braço com Nelfinavir.
A Figura a seguir exibe as estimativas de Kaplan-Meier do tempo para a falha de tratamento no Estudo 863. O tempo até a falha de tratamento foi definido como o menor tempo para que o paciente experimentasse falha virológica (2 valores consecutivos de RNA HIV demonstrando rebote, acima de 400 cópias/mL), um novo evento CDC de Classe C ou descontinuação prematura do estudo.
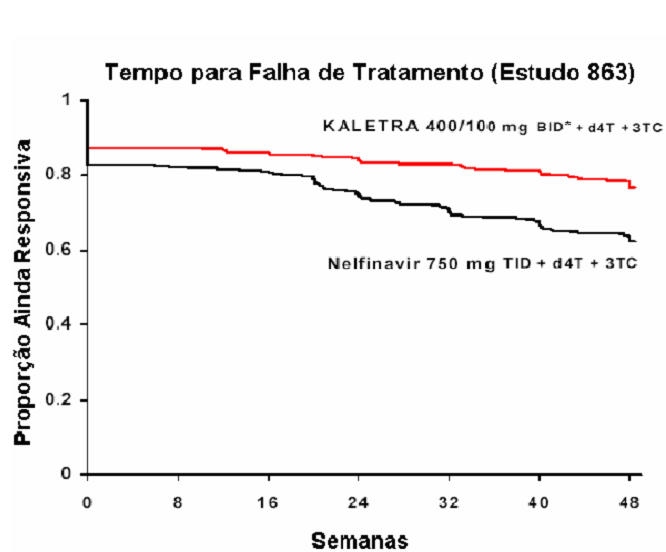
*BID - Bis in die ou duas vezes ao dia.
Estudo M97-720: Lopinavir + Ritonavir cápsulas duas vezes ao dia + Estavudina + Lamivudina
O Estudo M97-720 foi um ensaio randomizado, cego e multicêntrico avaliando o tratamento com Lopinavir + Ritonavir cápsulas em três diferentes doses (Grupo I: 200/100 mg duas vezes ao dia e 400/100 mg duas vezes ao dia; Grupo II: 400/100 mg duas vezes ao dia e 400/200 mg duas vezes ao dia) mais Lamivudina (150 mg duas vezes ao dia) e Estavudina (40 mg duas vezes ao dia), em 100 pacientes.
Todos os pacientes foram convertidos a Lopinavir + Ritonavir para o estudo de forma aberta na concentração de 400/100 mg duas vezes ao dia entre as Semanas 48 e 72 do estudo. Os pacientes tinham uma média de idade de 35 anos (faixa: 21 a 59), 70% eram caucasianos e 96% eram do sexo masculino.
A contagem celular média basal de CD4 era de 338 células/mm³ (faixa: 3 a 918 células/mm³) e o nível plasmático médio basal de RNA HIV-1 era de 4,9 log10 cópias/mL (faixa: 3,3 a 6,3 log10 cópias/mL).
Ao longo das 360 semanas de tratamento no Estudo 720, a proporção de pacientes com RNA HIV < 400 cópias/mL (< 50 cópias/mL) foi de 61% (59%) [n=100] e o aumento médio correspondente na contagem de células CD4 foi de 501 células/mm³. Trinta e nove pacientes (39%) descontinuaram o estudo, incluindo 15 (15%) descontinuações por eventos adversos e uma (1%) morte.
Dezoito pacientes demonstraram perda da resposta virológica (2 valores plasmáticos consecutivos para o RNA HIV-1 acima de 400 cópias/mL, um valor de rebote do RNA do HIV-1 seguido por descontinuação, ou falha em atingir o RNA HIV-1 <400 cópias/mL).
Análise genotípica de isolados virais foi conduzida nestes pacientes e em 10 pacientes adicionais com valores de RNA HIV-1 isolados > 400 cópias/ml após a semana 24. Os resultados de 19 pacientes não mostraram mutações primárias ou ativas na protease (aminoácidos nas posições 8, 30, 32, 36, 47, 48, 50, 82, 84 e 90) ou resistência fenotípica a inibidor de protease.
Estudo M05-730: Lopinavir + Ritonavir comprimidos uma vez a dia + Tenofovir DF + emtricitabina comparado com Lopinavir + Ritonavir duas vezes ao dia + Tenofovir DF + entricitabina
O Estudo M05-730 foi um ensaio randomizado, aberto e multicêntrico comparando o tratamento de Lopinavir + Ritonavir 800/200 mg uma vez ao dia com tenofovir DF e Emtricitabina com Lopinavir + Ritonavir 400/100 mg duas vezes ao dia mais tenofovir DF e entricitabina em 664 pacientes sem tratamento antirretroviral prévio (naive).
Os pacientes foram randomizados na razão 1:1 para receber Lopinavir + Ritonavir 800/200mg uma vez ao dia (n=333) ou Lopinavir + Ritonavir 400/100 mg duas vezes ao dia (n=331).
Uma estratificação adicional foi feita em cada grupo na proporção 1:1 (comprimidos versus cápsulas). Os pacientes que receberam cápsulas foram transferidos para a formulação comprimidos na oitava semana e mantidos na posologia da randomização. Os pacientes receberam 200 mg de emtricitabina e 300 mg de tenofovir DF uma vez ao dia. A média de idade dos pacientes foi de 39 anos (faixa: 19 a 71 anos); 75% eram caucasianos e 78% do sexo masculino.
A contagem celular média basal de CD4 era de 216 células/mm³ (faixa: 20 a 775 células/mm³) e o nível plasmático médio basal de RNA HIV-1 era de 5,0 log10 cópias/mL (faixa: 1,7 a 7,0 log10 cópias/mL).
A resposta ao tratamento e os resultados do tratamento randomizado na semana 48 estão presentes na tabela a seguir:
|
Resultados do Tratamento Randomizado até a Semana 48 (Estudo 730) |
||
|
Resultado |
Lopinavir + Ritonavir uma vez ao dia + TDF + FTC (N=333) |
Lopinavir + Ritonavir duas vezes ao dia + TDF + FTC (N=331) |
|
Respondedores1 |
78% | 77% |
|
Falha virológica2 |
10% | 8% |
|
Rebote2 |
5% | 5% |
|
Nunca suprimiu até a Semana 482 |
5% | 3% |
|
Morte |
1% | < 1% |
|
Descontinuou por evento adverso |
4% | 3% |
|
Descontinuou por outras razões3 |
8% | 11% |
1 Pacientes atingiram e mantiveram RNA HIV-1 confirmado de <50 cópias/mL até a Semana 48.
2 Inclui as situações confirmadas de rebotes virais e falhas para atingir <50 cópias/mL, até a Semana 48.
3 Inclui perda de acompanhamento, retirada do paciente, não-aderência, violação do protocolo e outras razões.
Até a Semana 48 de terapia, 78% dos pacientes do braço de Lopinavir + Ritonavir uma vez ao dia e 77% do braço Lopinavir + Ritonavir duas vezes ao dia atingiram e mantiveram RNA HIV-1 <50 cópias/mL (95% do intervalo de confiança e diferença de 5,9% para 6,8%).
A contagem celular média de CD4 aumentou na Semana 48 foi de 186 células/mm³ no braço Lopinavir + Ritonavir uma vez ao dia e 198 células/mm³ no braço Lopinavir + Ritonavir duas vezes ao dia.
Pacientes com Tratamento Antirretroviral Prévio
Atividade Antiviral de Lopinavir + Ritonavir em Pacientes com Terapia Prévia com Inibidor de Protease
A relevância clínica da sensibilidade reduzida in vitro a lopinavir foi examinada pela avaliação da resposta virológica à terapia com Lopinavir + Ritonavir com relação ao genótipo e fenótipo viral basal, em 56 pacientes naive para NNRTI* com RNA HIV superior a 1.000 cópias/mL, a despeito de terapia prévia com pelo menos 2 inibidores de protease, selecionados entre Nelfinavir, Indinavir, Saquinavir e Ritonavir (Estudo M98-957).
Nesse estudo, os pacientes foram inicialmente randomizados para receber uma de duas doses de Lopinavir + Ritonavir , em combinação com efavirenz e inibidores nucleosídeos da transcriptase reversa.
*NNRTI = Non-Nucleoside Reverse Transcriptase Inhibitor ou Inibidor Não-nucleosídeo da Transcriptase Reversa.
Os valores para a EC50 de lopinavir contra os 56 isolados virais basais, variaram de números entre 0,5 a 96 vezes mais elevados que a EC50 contra o tipo selvagem de HIV. Cinquenta e cinco por cento (31/56) destes isolados basais exibiram uma sensibilidade a lopinavir reduzida mais que 4 vezes. Estes 31 isolados tiveram uma redução média na sensibilidade a lopinavir de 27,9 vezes.
Após 48 semanas de tratamento com Lopinavir + Ritonavir , efavirenz e inibidores nucleosídeos da transcriptase reversa, um valor plasmático para o RNA HIV menor que, ou igual a, 400 cópias/mL, foi observado em 93% (25/27), 73% (11/15) e 25% (2/8) dos pacientes com sensibilidade a lopinavir reduzida, menos que ou igual a 10 vezes; mais que 10 e menos que 40 vezes e mais que, ou igual a, 40 vezes àquela da linha basal, respectivamente.
A sensibilidade ao lopinavir foi determinada pela tecnologia de fenotipagem recombinante desenvolvida pela Virologic; o genótipo também foi determinado pela Virologic. Um valor plasmático para o RNA HIV menor que, ou igual, a 50 cópias/mL, foi observado em 81% (22/27), 60% (9/15) e 25% (2/8) nos grupos de pacientes acima, respectivamente.
Há dados insuficientes neste momento para identificar os padrões de mutação associados ao lopinavir nos isolados provenientes de pacientes sob tratamento com Lopinavir + Ritonavir. Outros estudos são necessários para avaliar a associação entre padrões mutacionais específicos e taxas de resposta virológica.
Estudo M98-888: Lopinvair + Ritonavir Lopinavir + Ritonavir cápsulas duas vezes ao dia + Nevirapina + ITRNs em comparação a outros inibidores de protease selecionados pelo Investigador + Nevirapina + ITRNs
O Estudo 888 foi um ensaio randomizado, aberto, multicêntrico comparando o tratamento com Lopinavir + Ritonavir cápsulas (400/100 mg duas vezes ao dia) + Nevirapina e inibidores nucleosídeos da transcriptase reversa versus outros inibidores de protease selecionados pelo Investigador + Nevirapina e inibidores nucleosídeos da transcriptase reversa, em 288 pacientes que já haviam recebido esquema com inibidor de protease único e que nunca haviam recebido inibidores não-nucleosídeos da transcriptase reversa (NNRTI).
Os pacientes tinham uma média de idade de 40 anos (faixa: 18 a 74), 68% eram caucasianos e 86% eram do sexo masculino.
A contagem celular basal média de CD4 foi de 322 células/mm³ (faixa: 10 a 1.059 células/mm³) e o nível plasmático médio de RNA HIV-1 à linha basal era de 4,1 log10 cópias/mL (faixa: 2,6 a 6,0 log10 cópias/mL).
A resposta ao tratamento e os resultados dos tratamentos randomizados até a Semana 48 são apresentados na figura e na tabela a seguir, respectivamente:

* Ensaio AMPLICOR HIV-1 MONITOR, da Roche.
† Responsivos a cada visita, são os pacientes que habiam atingido e mantido uma concentração de RNA do HIV -1 <400 cópias/ML sem descontinuação naquela visita.
|
Resultados do Tratamento Randomizado até a Semana 48 (Estudo 888) |
||
|
Resultado |
Lopinavir + Ritonavir + Nevirapina+ ITRNs (N=148) |
Inibidor(es) de protease selecionado(s) pelo Investigador + Nevirapina+ ITRNs (N=140) |
|
Respondedores*1 |
57% | 33% |
|
Falha virológica2 |
24% | 41% |
|
Rebote2 |
11% | 19% |
|
Nunca suprimiu até a Semana 482 |
13% | 23% |
|
Morte |
1% | 2% |
|
Descontinuou por evento adverso |
5% | 11% |
|
Descontinuou por outras razões3 |
14% | 13% |
* Corresponde às taxas na Semana 48 da Figura acima.
1 Pacientes atingiram e mantiveram RNA HIV confirmado de <400 cópias/mL, até a Semana 48.
2 Inclui as situações confirmadas de rebotes virais e falhas para atingir <400 cópias/mL, até a Semana 48.
3 Inclui perda de acompanhamento, retirada do paciente, não-aderência, violação do protocolo e outras razões.
Estudo M97-765: Lopinavir + Ritonavir cápsulas duas vezes ao dia + Nevirapina + NRTIs
O Estudo M97-765 foi um ensaio randomizado, cego, multicêntrico, avaliando tratamento com Lopinavir + Ritonavir cápsulas em 2 níveis de dose (400/100 mg duas vezes ao dia e 400/200 mg duas vezes ao dia) mais Nevirapina(200 mg duas vezes ao dia) e 2 NRTIs em 70 pacientes que já haviam recebido esquema com inibidor de protease único, que nunca haviam recebido inibidores não-nucleosídeos de transcriptase reversa (NNRTI).
Os pacientes tinham uma média de idade de 40 anos (faixa: 22 a 66 anos), 73% eram caucasianos e 90% do sexo masculino. A contagem celular média basal de CD4 era 372 células/mm³ (faixa: 72 a 807 células/mm³) e a concentração plasmática basal média de RNA do HIV-1 era de 4,0 log10 cópias/mL (faixa: 2,9 a 5,8 log10 cópias/mL).
Ao longo de 144 semanas de tratamento no Estudo 765, a proporção de pacientes com RNA HIV <400 (<50) cópias/mL foi de 54% (50%) [n=70] e o correspondente aumento médio na contagem de células CD4 foi de 212 células/mm³. 27 pacientes (39%) descontinuaram o estudo, incluindo 9 (13%) descontinuações secundárias a eventos adversos e 2 (3%) óbitos.
Estudo M06-802 Lopinavir + Ritonavir comprimidos 800/200 mg uma vez ao dia versus Lopinavir + Ritonavir comprimidos 400/100 mg duas vezes ao dia e coadministrados com inibidores da Transcriptase Reversa Análogos de Nucleosídeo/Nucleotídeo em pacientes infectados pelo HIV-1 com experiência aos antirretrovirais
O estudo M06-802 foi um ensaio randomizado, aberto, comparando-se a segurança, tolerabilidade e atividade antirretroviral de Lopinavir + Ritonavir uma vez ao dia e duas vezes ao dia em 599 pacientes com carga viral detectável enquanto recebiam a terapia antirretroviral.
Os pacientes foram randomizados na razão 1:1 para receber Lopinavir + Ritonavir 800/200mg uma vez ao dia (n=300) ou Lopinavir + Ritonavir 400/100 mg duas vezes ao dia (n=299).
Os pacientes recebiam, pelo menos, dois inibidores da transcriptase reversa análogos de nucleosídeos/nucleotídeos selecionados pelo investigador. A média de idade dos pacientes foi de 41 anos (faixa: 21 a 73 anos); 51% eram caucasianos e 66% do sexo masculino.
A contagem celular média basal de CD4 era de 254 células/mm³ (faixa: 4 a 952 células/mm³) e o nível plasmático médio basal de RNA HIV-1 era de 4,3 log10 cópias/mL (faixa: 1,7 a 6,6 log10 cópias/mL).
A resposta ao tratamento e os resultados dos tratamentos randomizados até a Semana 48 são apresentados na tabela a seguir:
|
Resultados do Tratamento Randomizado até a Semana 48 (Estudo 802) |
||
|
Resultado |
Lopinavir + Ritonavir uma vez ao dia + ITRNs |
Lopinavir + Ritonavir duas vezes ao dia + ITRNs |
|
Respondedores*1 |
55% | 52% |
|
Falha virológica2 |
25% | 28% |
|
Rebote2 |
12% | 14% |
|
Nunca suprimiu até a Semana 482 |
13% | 14% |
|
Morte |
1% | 1% |
|
Descontinuou por evento adverso |
4% | 6% |
|
Descontinuou por outras razões3 |
15% | 14% |
1 Pacientes atingiram e mantiveram RNA HIV-1 confirmado de <50 cópias/mL até a Semana 48.
2 Inclui as situações confirmadas de rebotes virais e falhas para atingir <50 cópias/mL, até a Semana 48.
3 Inclui perda de acompanhamento, retirada do paciente, não-aderência, violação do protocolo e outras razões.
Uso Pediátrico
Estudo M98-9405: Um estudo aberto de fase I/II de ABT-378/ritonavir em combinação com inibidores de transcriptase reversa em pacientes pediátricos infectados com HIV
O Estudo M98-940 foi um ensaio aberto e multicêntrico avaliando o perfil farmacocinético, tolerabilidade, segurança e eficácia de Lopinavir + Ritonavir solução oral contendo lopinavir 80 mg/mL e ritonavir 20 mg/mL, em 100 pacientes pediátricos naive (44%) e que já haviam sido tratados (56%) com algum antirretroviral. Todos os pacientes eram naive para inibidor não-nucleosídeo da transcriptase reversa.
Os pacientes foram randomizados ou para 230 mg lopinavir/57,5 mg Ritonavir por m² ou para 300 mg lopinavir/75 mg Ritonavir por m². Os pacientes naive também receberam Lamivudina e Estavudina. Os pacientes que já haviam sido tratados receberam Nevirapinae até 2 inibidores nucleosídeos da transcriptase reversa.
A segurança, eficácia e perfis farmacocinéticos dos 2 regimes posológicos foram avaliados após 3 semanas de tratamento em cada paciente. Após a análise destes dados, todos os pacientes continuaram sob uma dose de 300 mg de lopinavir/75 mg de ritonavir por m².
Os pacientes tinham uma média de idade de 05 anos (faixa: 06 meses a 12 anos) com 14% deles sendo menores que 02 anos de idade. A contagem celular basal média de CD4 era de 838 células/mm³ e a concentração plasmática média basal de RNA HIV-1 era de 4,7 log10 cópias/mL.
Ao longo das 48 semanas de tratamento, a proporção de pacientes que atingiu e manteve uma concentração plasmática de RNA HIV <400 cópias/mL foi de 80% entre os pacientes naive para antirretrovirais e de 71% entre os pacientes experientes com antirretrovirais.
O aumento médio desde a linha basal na contagem celular de CD4 foi de 404 células/mm³ para os pacientes naive para antirretrovirais e de 284 células/mm³ para os pacientes experimentados, ao longo da 48 semanas.
Descontinuações prematuras foram notadas em 2 (2%) indivíduos antes da Semana 48. Uma destas foi considerada pelo Investigador como “não-relacionada” ao fármaco do estudo, a segunda como “possivelmente” relacionada ao fármaco do estudo.
A seleção de dose para pacientes com 06 meses a 12 anos de idade baseou-se nos resultados a seguir. O regime com 230/57,5 mg/m² duas vezes ao dia sem Nevirapinae o regime com 300/75 mg/m² duas vezes ao dia com Nevirapinaforneceram concentrações plasmáticas de lopinavir similares àquelas obtidas em pacientes adultos recebendo o regime de 400/100 mg duas vezes ao dia (sem nevirapina).
KONCERT/PENTA 18: Estudo randomizado de farmacocinética, segurança e eficácia do uso de Lopinavir + Ritonavir comprimidos duas vezes por dia versus uma vez por dia doseados por peso como parte da combinação da terapia antirretroviral em pacientes pediátricos/crianças, da rede européia. infectados pelo HIV-1 para o tratamento da AIDS
KONCERT/PENTA 18 é um estudo multicêntrico prospectivo, randomizado e aberto que avaliou o perfil farmacocinético, de eficácia e de segurança de dosagem duas vezes por dia versus uma vez por dia de Lopinavir + Ritonavir comprimidos 100/25 mg dosados por peso como parte da terapia antiretroviral combinada (cART) em crianças infectadas com HIV-1 virologicamente suprimidas (n = 173).
As crianças foram elegíveis quando elas tinham idade < 18 anos, ≥ 15 kg de peso, recebendo cART que incluia Lopinavir + Ritonavir , ácido ribonucleico (RNA) de HIV-1 < 50 cópias/mL por pelo menos 24 semanas e capazes de engolir comprimidos.
Na semana 24, a eficácia e segurança com a dosagem de 2 vezes por dia (n=87) na população pediátrica que recebeu Lopinavir + Ritonavir comprimidos 100/25 mg foi consistente com os estudos de eficácia e a segurança em estudos pediátricos e em adultos anteriores, usando Lopinavir + Ritonavir duas vezes por dia.
Características Farmacológicas
Geral
Lopinavir + Ritonavir é uma formulação combinada de lopinavir e ritonavir. O Lopinavir é um inibidor das proteases do HIV-1 e do HIV-2.
O Ritonavir presente na formulação de Lopinavir + Ritonavir inibe o metabolismo mediado pelo CYP3A (citocromo P450 3A) do lopinavir, proporcionando, deste modo, níveis plasmáticos maiores de lopinavir.
Farmacodinâmica
Mecanismo de ação:
Lopinavir é um inibidor das proteases do HIV-1 e HIV-2. A inibição da protease previne a clivagem da poliproteína gag-pol levando à formação de um vírus imaturo, não infeccioso.
O tempo de início de ação não é aplicável para Lopinavir + Ritonavir uma vez que este medicamento é administrado cronicamente. Com uma dose de 400/100 mg, duas vezes ao dia, as concentrações mínimas de lopinavir são superiores a EC50 do vírus de tipo selvagem (0,07 mcg / mL) por um fator maior que 50, sugerindo que as concentrações de lopinavir são mantidas para a eficácia em todo o intervalo de doses.
Atividade antiviral in vitro:
A atividade antiviral de lopinavir foi avaliada in vitro contra cepas laboratoriais e clínicas do HIV em linhagens de células linfoblásticas e isolados clínicos do HIV em linfócitos periféricos de sangue, infectados agudamente.
Na ausência do soro humano, a CE50 (concentração eficaz 50%) de lopinavir contra cinco cepas laboratoriais diferentes de HIV-1 variou de 10 a 27 nM (0,006 a 0,017 mcg/mL, 1 mcg/mL equivalente a 1,6 μM) e variou de 4 a 11 nM (0,003 a 0,007 mcg/mL) contra vários isolados clínicos de HIV-1 (n=6).
Na presença de 50% de soro humano, a CE50 do Lopinavir contra essas 5 cepas laboratoriais variou de 65 a 289 nM (0,04 a 0,18 mcg/mL), representando uma atenuação de 7 a 11 vezes. Estudos sobre a atividade da combinação de substâncias com Lopinavir e outros inibidores de proteases ou inibidores da transcriptase reversa não foram completados.
Resistência:
Isolados de HIV-1 com sensibilidade reduzida ao Lopinavir foram selecionados in vitro. A presença do ritonavir não parece influenciar a seleção de vírus resistentes ao lopinavir in vitro.
Ainda não foi caracterizada a seleção de resistência ao Lopinavir + Ritonavir no tratamento antirretroviral em pacientes que nunca receberam antirretrovirais. Em um estudo de fase III envolvendo 653 pacientes que nunca haviam recebido tratamento antirretroviral (Estudo 863), foram analisados os isolados virais plasmáticos de cada paciente em tratamento que apresentou HIV plasmático maior do que 400 cópias/mL nas semanas 24, 32, 40 e/ou 48.
Nenhuma evidência de resistência genotípica ou fenotípica a Lopinavir + Ritonavir foi observada em 37 pacientes avaliáveis tratados com Lopinavir + Ritonavir (0%).
Evidência de resistência genotípica a nelfinavir, definida como a presença de mutação D30N e/ou L90M na protease do HIV, foi observada em 25/76 (33%) dos pacientes avaliáveis tratados com nelfinavir. A seleção de resistência a Lopinavir + Ritonavir em pacientes pediátricos que nunca haviam recebido tratamento antirretroviral (Estudo 940), parece ser consistente com aquela vista em pacientes adultos (Estudo 863).
Resistência ao Lopinavir + Ritonavir foi observada em pacientes que fizeram uso prévio de outros inibidores de protease antes da terapia com Lopinavir + Ritonavir.
Em estudos de Fase II com 227 pacientes que nunca haviam recebido tratamento com antirretroviral e multiexperimentados com inibidores de protease, 4 de 23 pacientes com carga viral maior que 400 cópias/mL após tratamento com Lopinavir + Ritonavir por 12 a 100 semanas, mostraram sensibilidade significativamente reduzida ao lopinavir em comparação aos correspondentes isolados virais basais.
Três desses pacientes haviam recebido previamente tratamento com um único inibidor de protease (Nelfinavir, Indinavir ou Saquinavir) e um paciente havia recebido tratamento com múltiplos inibidores de proteases (indinavir, saquinavir, ritonavir).
Os quatro pacientes apresentavam pelo menos 4 mutações associadas à resistência ao inibidor de protease imediatamente antes do tratamento com Lopinavir + Ritonavir.
Depois da replicação viral, todos os isolados desses pacientes continham mutações adicionais, algumas das quais são sabidamente associadas à resistência a inibidores de protease. Entretanto, os dados são insuficientes até o momento para identificar padrões de mutação associada ao lopinavir em isolados de pacientes em tratamento com Lopinavir + Ritonavir. A avaliação desses padrões de mutação está em estudo.
Resistência cruzada:
Estudos pré-clínicos:
Entre os inibidores de proteases foram observados vários graus de resistência cruzada. Foi determinada a atividade in-vitro do lopinavir contra isolados clínicos de pacientes previamente tratados com um único inibidor de protease.
Isolados que apresentaram sensibilidade reduzida acima de 4 vezes, ao nelfinavir (n=13) e saquinavir (n=4), apresentaram sensibilidade reduzida abaixo de 4 vezes ao lopinavir.
Isolados com sensibilidade reduzida acima de 4 vezes ao indinavir (n=16) e ritonavir (n=3) apresentaram a média de redução de sensibilidade de 5,7 e 8,3 vezes em relação ao lopinavir, respectivamente. Isolados de pacientes previamente tratados com dois ou mais inibidores de protease apresentaram maior redução da sensibilidade ao Lopinavir, conforme descrito no item “Resultados de Eficácia: Atividade antiviral do Lopinavir + Ritonavir em pacientes previamente tratados com inibidores de protease”.
Resistência cruzada durante tratamento com Lopinavir + Ritonavir:
Dispõe-se de pouca informação a respeito da resistência cruzada de vírus selecionados durante o tratamento com Lopinavir + Ritonavir.
Isolados de quatro pacientes tratados previamente com um ou mais inibidores de protease que desenvolveram resistência fenotípica ao Lopinavir durante o tratamento com Lopinavir + Ritonavir , tanto permaneceram com a resistência cruzada ou desenvolveram resistência cruzada ao Ritonavir, Indinavir e Nelfinavir.
Todos os vírus permaneceram completamente sensíveis ou apresentaram sensibilidade modestamente reduzida ao amprenavir (resistência de até 8,5 vezes concomitante a 99 vezes ao lopinavir). Os isolados virais de dois indivíduos sem tratamento prévio ao saquinavir permaneceram completamente sensíveis ao saquinavir.
Correlatos genotípicos de resposta virológica reduzida em pacientes com tratamento antirretroviral prévio iniciando terapia combinada com Lopinavir + Ritonavir:
A resposta virológica a Lopinavir + Ritonavir mostrou-se afetada pela presença de três ou mais das seguintes substituições aminoácidas na protease basal: L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V.
A tabela abaixo mostra a resposta virológica de 48 semanas (RNA HIV menor que 400 cópias/ mL) de acordo com as mutações da resistência de inibição de protease basal acima mencionadas nos Estudos 888 e 765 e Estudo 957.
Resposta virológica (RNA HIV menor que 400 cópias/ mL) na Semana 48 pela susceptibilidade basal de Lopinavir + Ritonavir e pelo número de substituições de protease associado à resposta a Lopinavir + Ritonavir 1:
 1 Substituições consideradas na análise incluem L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V.
1 Substituições consideradas na análise incluem L10F/I/R/V, K20M/N/R, L24I, L33F, M36I, I47V, G48V, I54L/T/V, V82A/C/F/S/T e I84V.
2 43% indinavir, 42% Nelfinavir, 10% ritonavir, 15% saquinavir.
3 41% indinavir, 38% Nelfinavir, 4% ritonavir, 16% saquinavir.
4 86% indinavir, 54% nelfinavir, 80% ritonavir, 70% saquinavir.
Farmacocinética
As propriedades farmacocinéticas do lopinavir coadministrado com ritonavir foram avaliadas em voluntários adultos sadios e em pacientes infectados pelo HIV; e não foram observadas diferenças substanciais entre os 2 grupos. O Lopinavir é completamente metabolizado pelo CYP3A. O Ritonavir inibe o metabolismo do lopinavir, aumentando, deste modo, os níveis plasmáticos de lopinavir.
Nos estudos, a administração de Lopinavir + Ritonavir 400/100 mg, duas vezes ao dia, proporcionou concentrações plasmáticas médias no estado de equilíbrio (steady-state) 15 a 20 vezes maiores do que aquelas com ritonavir, em pacientes infectados pelo HIV.
Os níveis plasmáticos de ritonavir são menos de 7% daqueles obtidos após a administração de 600 mg de ritonavir duas vezes ao dia. A CE50 antiviral do lopinavir in vitro é aproximadamente 10 vezes menor do que a do Ritonavir. Deste modo, a atividade antiviral de Lopinavir + Ritonavir é devida ao Lopinavir.
A Figura a seguir mostra as concentrações plasmáticas médias obtidas no estado de equilíbrio de lopinavir e ritonavir, após Lopinavir + Ritonavir 400/100 mg duas vezes ao dia com alimentação por 3 semanas; dado este proveniente de um estudo farmacocinético em indivíduos adultos infectados com HIV (n=19).
As concentrações plasmáticas de lopinavir e ritonavir após a administração de 2 comprimidos de 200/50 mg, são equivalentes àquelas de 3 cápsulas de 133/33 mg sob condição de não-jejum, com menos variabilidade farmacocinética.

Absorção:
Em um estudo de farmacocinética com 19 pacientes infectados pelo HIV, a administração múltipla de Lopinavir + Ritonavir 400/100 mg, duas vezes ao dia, com alimentos, durante 3 semanas produziu uma concentração plasmática pico (Cmáx) (média ± desvio padrão) de 9,8 ± 3,7 μg/mL, que ocorreu aproximadamente 4 horas após a administração.
A concentração média no estado de equilíbrio antes da dose matinal foi de 7,1 ± 2,9 μg/mL e a Cmin no intervalo da dose foi de 5,5 ± 2,7 μg/mL. A AUC do lopinavir durante um período de administração de 12 horas foi em média de 92,6 ± 36,7 μg.h/mL.
A biodisponibilidade absoluta de lopinavir em fórmula combinada com ritonavir não foi determinada em humanos. Sob condições pós–prandiais (500 kcal/ 25% proveniente de gordura), as concentrações de lopinavir foram similares na administração de Lopinavir + Ritonavir cápsulas e solução oral.
Na administração de Lopinavir + Ritonavir em jejum, a AUC e a Cmáx do lopinavir foram 22% menores para o Lopinavir + Ritonavir solução oral em relação às cápsulas.
Efeitos da alimentação sobre a absorção oral:
Lopinavir + Ritonavir cápsulas e solução oral foram bioequivalentes em condições pós-prandiais (refeição com teor moderado de gordura).
A administração de uma dose única de 400/100 mg de Lopinavir + Ritonavir cápsulas com uma refeição com teor moderado de gordura (500-682 Kcal, 23 a 25% de calorias provenientes de gordura) foi associada com um aumento médio de 48% e 23% na AUC e na Cmáx do lopinavir, respectivamente, em relação ao jejum.
Para Lopinavir + Ritonavir solução oral os aumentos correspondentes na AUC e na Cmáx de lopinavir foram de 80% e 54%, respectivamente. A administração de Lopinavir + Ritonavir com uma refeição rica em gorduras (872 Kcal, 56% provenientes de gordura) aumentou a AUC e a Cmáx do lopinavir em 97% e 43%, respectivamente, para as cápsulas e 130% e 56%, respectivamente, para a solução oral.
Para aumentar a biodisponibilidade e minimizar a variabilidade, Lopinavir + Ritonavir deve ser tomado com as refeições.
Distribuição:
No estado de equilíbrio, lopinavir está aproximadamente 98-99% ligado a proteínas plasmáticas. O lopinavir se liga tanto à alfa-1 glicoproteína ácida quanto à albumina, no entanto, tem maior afinidade pela alfa-1 glicoproteína ácida.
No estado de equilíbrio, a ligação de lopinavir a proteínas permanece constante em todas as concentrações observadas após a administração de Lopinavir + Ritonavir , 400/100 mg, duas vezes ao dia, e é similar em voluntários sadios e pacientes HIV- positivos.
Metabolismo:
Experiências in vitro com microssomas hepáticos humanos indicam que lopinavir sofre principalmente metabolismo oxidativo. Lopinavir é extensamente metabolizado pelo sistema citocromo P450 hepático, quase que exclusivamente pela isoenzima CYP3A. O ritonavir é um potente inibidor do CYP3A, inibindo o metabolismo do lopinavir e, deste modo, aumentando os níveis plasmáticos de lopinavir.
Um estudo com Lopinavir marcado com 14C em humanos mostrou que 89% da radioatividade plasmática após uma administração única de Lopinavir + Ritonavir 400/ 100 mg foi decorrente da droga-mãe.
Pelo menos 13 metabólitos oxidativos de lopinavir foram identificados no ser humano. O Ritonavir mostrou induzir enzimas metabólicas, resultando na indução de seu próprio metabolismo. As concentrações de Lopinavir antes da administração diminuem com o tempo com a administração múltipla, estabilizando após cerca de 10 a 16 dias.
Eliminação:
Após uma dose de 400/100 mg de Lopinavir/Ritonavir marcados com 14C, aproximadamente 10,4 ± 2,3% e 82,6 ± 2,5% de uma dose administrada de lopinavir marcado com 14C pode ser encontrado na urina e nas fezes, respectivamente, depois de 8 dias. Lopinavir inalterado correspondeu a aproximadamente 2,2% e 19,8% da dose administrada na urina e fezes, respectivamente. Após múltiplas doses, menos de 3% da dose de lopinavir é excretada inalterada pela urina. A depuração aparente (CL/F) do lopinavir é 5,98 ± 5,75 L/h (média ± desvio padrão, n= 19).
Administração uma vez ao dia:
A farmacocinética de Lopinavir + Ritonavir comprimido administrado uma única vez ao dia foi avaliada em pacientes sem tratamento antirretroviral prévio (naive). Lopinavir + Ritonavir (Lopinavir/Ritonavir) 800mg/200mg foi administrado em combinação com emtricitabina 200mg e tenofovir DF 300mg como parte do esquema posológico de uma vez ao dia.
Múltiplas doses de Lopinavir + Ritonavir comprimido 800mg/200mg uma vez ao dia por 2 semanas sem restrição alimentar (n=16) produziram uma média ± desvio padrão da Cmax de 14,8 ± 3,5μg/mL, a qual ocorreu aproximadamente 6 horas após a administração. A concentração mínima no estado de equilíbrio de lopinavir antes da administração matinal foi de 5,5 ± 5,4 μg/mL e a concentração mínima dentro do período entre doses foi de 3,2 ± 3,4 μg/mL. A AUC de lopinavir em um intervalo de 24 horas de administração foi em média 206,5 ± 89,7 μg•h/mL.
Efeitos no Eletrocardiograma:
O intervalo QTcF foi avaliado em um estudo controlado cruzado, randomizado, placebo e ativo (moxifloxacina 400 mg/uma vez ao dia), com 39 adultos sadios, com 10 medidas durante 12 horas no Dia 3. A média de diferença máxima (intervalo de confiança superior a 95%) no QTcF do placebo foi de 3,6 (6,3) mseg e 13,1 (15,8) mseg para Lopinavir/Ritonavir 400/100 mg duas vezes ao dia e supraterapêutica de 800/200 mg duas vezes ao dia, respectivamente.
Os dois regimes resultaram em exposições no Dia 3 de, aproximadamente, 1,5 a 3 vezes maior que as observadas com as doses recomendadas nas terapias de dose única diária e duas doses diárias no estado de equilíbrio. Nenhum voluntário teve um aumento na QTcF > 60 mseg da baseline ou um intervalo QTcF que excedesse o limite clinicamente relevante de 500 mseg.
Um discreto prolongamento no intervalo PR também foi verificado em voluntários recebendo Lopinavir/Ritonavir durante o mesmo estudo no Dia 3. O intervalo PR máximo foi de 286 mseg e não houve bloqueio cardíaco de segundo ou terceiro graus em voluntários deste estudo.
Carcinogênese e mutagênese:
Estudos de longo prazo sobre carcinogênese com Lopinavir + Ritonavir em ratos revelaram a indução não-genotóxica, não-mitogênica de tumores hepáticos, geralmente considerados de baixo risco em humanos. Estudos de carcinogênese em ratos não se mostraram tumorigênicos.
O Lopinavir não se mostrou mutagênico ou clastogênico em uma bateria de ensaios in vitro, incluindo o teste de Ames de mutação bacteriana reversa, o teste de linfoma em camundongos e testes de aberração cromossômica em linfócitos humanos. Lopinavir + Ritonavir não foi mutagênico ou clastogênico em testes in vivo com micronúcleos de camundongos.
Qual a composição do Kaletra?
Cada mL de solução contém:
| Lopinavir |
80 mg |
| Ritonavir |
20 mg |
Excipientes: álcool etílico, xarope de milho com alto teor de frutose, propilenoglicol, água purificada, glicerol, povidona, sabor artificial e natural de baunilha, sabor artificial de algodão doce, aroma magnasweet, óleo de rícino hidrogenado polioxil 40, acessulfamo potássico, sacarina sódica, cloreto de sódio, óleo de hortelã, citrato de sódio, ácido cítrico e mentol.
Kaletra® (lopinavir/ritonavir) solução oral contém 42,4% de álcool (v/v) e 15,3% de propilenoglicol (p/v).
Cada comprimido revestido de Kaletra® (lopinavir/ritonavir) 200 mg/50 mg contém:
| Lopinavir |
200 mg |
| Ritonavir |
50 mg |
Excipientes: copovidona, laurato de sorbitana, dióxido de silício, estearilfumarato de sódio, hipromelose, dióxido de titânio, macrogol, hiprolose, talco, óxido de ferro amarelo e polissorbato 80.
Cada comprimido revestido de Kaletra® (lopinavir/ritonavir) 100 mg/25 mg contém:
| Lopinavir |
100 mg |
| Ritonavir |
25 mg |
Excipientes: copovidona, laurato de sorbitana, dióxido de silício, estearilfumarato de sódio, álcool polivinílico, dióxido de titânio, talco, macrogol e óxido de ferro amarelo.
Como devo armazenar o Kaletra?
Número de lote e datas de fabricação e validade: Vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Exclusivo solução oral: Kaletra® (lopinavir/ritonavir) solução oral deve ser conservado sob refrigeração (temperatura entre 2 e 8°C).
Exclusivo comprimido revestido: Kaletra® (lopinavir/ritonavir) deve ser armazenado em temperatura ambiente (temperatura entre 15 e 30°C).
Características físicas e organolépticas
Kaletra® (lopinavir/ritonavir) solução oral
Apresenta-se como uma solução amarelada, límpida e livre de partículas.
Kaletra® (lopinavir/ritonavir) comprimido 100 mg/25 mg
Apresenta-se como comprimido ovaloide de coloração amarelo claro.
Kaletra® (lopinavir/ritonavir) comprimido 200 mg/50 mg
Apresenta-se como comprimido oval de coloração amarela.
Antes de usar, observe o aspecto do medicamento. Caso ele esteja no prazo de validade e você observe alguma mudança no aspecto, consulte o farmacêutico para saber se poderá utilizá-lo.
Todo medicamento deve ser mantido fora do alcance das crianças.
Dizeres Legais do Kaletra
Solução oral / Comprimido revestido
M.S - 1.9860.0010
Farm. Resp.:
Joyce M. C. Camargo
CRF-SP nº 17.077
Importado por:
AbbVie Farmacêutica Ltda.
Av. Guido Caloi, 1935, 1º andar, Bloco C - São Paulo - SP
CNPJ: 15.800.545/0001-50
Venda sob prescrição médica.
Exclusivo solução oral
Fabricado por:
AbbVie Inc
N Waukegan Rd, North Chicago - EUA
Exclusivo comprimido revestido
Kaletra® (lopinavir/ritonavir) comprimidos revestidos 100 mg/25 mg
Fabricado e embalado por:
AbbVie Deutschland GmbH & Co. KG
Ludwigshafen - Alemanha
Kaletra® (lopinavir/ritonavir) comprimidos revestidos 200 mg/50 mg:
Fabricado e embalado por:
AbbVie Deutschland GmbH & Co. KG
Ludwigshafen – Alemanha
Ou
Fabricado por:
AbbVie Deutschland GmbH & Co. KG
Ludwigshafen - Alemanha
E
Embalado por:
AbbVie Inc
N. Waukegan Road
North Chicago - EUA
Doenças relacionadas
Quer saber mais?
Consulta também aBula do Lopinavir + Ritonavir
O conteúdo desta bula foi extraído manualmente da bula original, sob supervisão técnica do(a) farmacêutica responsável: Karime Halmenschlager Sleiman (CRF- PR 39421). Última atualização: 18 de Março de 2025.